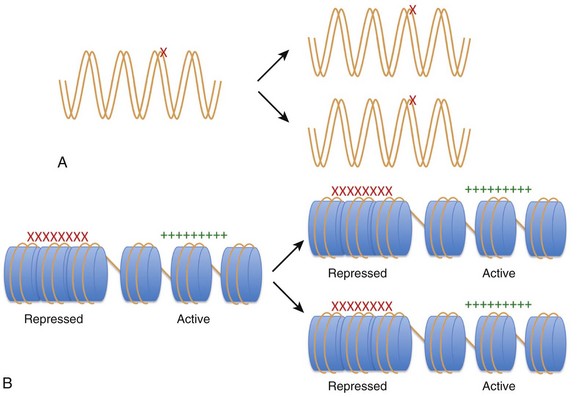
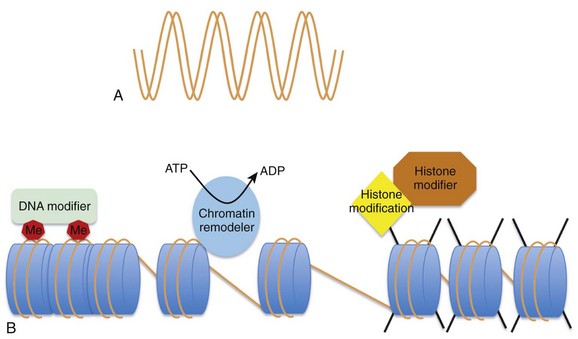
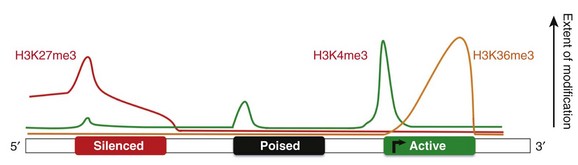
24 Genetic mutations are well-recognized causes of heritable phenotypic shifts. The completion of the Human Genome Project and technical advances that allow for high-throughput sequencing have advanced the exploration for genetic signatures that associate with and, perhaps, cause disease states. However, it is very possible that many disease states result from environmental influences that affect the expression or repression of different genes without altering the underlying DNA sequence. By definition, an epigenetic phenomenon is a heritable alteration in phenotype without an underlying DNA mutation. Epigenetic phenomena regulate gene expression profiles by controlling genes in a binary manner such that genes are actively expressed (i.e., “on”) or repressed (i.e., “off”). In addition, true epigenetic changes that occur as a result of a stimulus should be heritable and stable, even in the subsequent absence of that stimulus. Although the concept of epigenetics seems simple, it differs distinctly from the classic notion that only mutations at the DNA level can cause heritable phenotypic shifts (Figure 24-1). Clinicians and scientists are exploring epigenetic mechanisms and their contribution to the development of disease states, because these phenomena provide a rational mechanistic explanation for how environmental factors can alter phenotypes in a heritable manner in the absence of DNA mutations. Figure 24-1 Heritability of phenotypic changes at the level of DNA (A) and by epigenetic mechanisms (B). A, The genetic code is defined by the DNA template (orange lines). In this model, DNA mutations (red X) can alter the expression of genes and the resultant phenotype. DNA replication results in mutations being passed from a mother cell to her progeny. B, The process by which epigenetic mechanisms can regulate gene expression profiles in a heritable manner without mutating the underlying DNA. The expression of genes can be regulated by interactions between nucleosome components (blue cylinders) and the DNA (orange strand) such that certain genes are repressed (red X) and certain genes are active (green +). The DNA and the nucleosome components are both heritable so that the active and repressed gene patterns are similar in the mother cells and daughter progeny. Early work in Drosophila species development revealed that the homeotic genes define segment identity and the position of appendages along the body axes. Thus, developmental biologists became interested in mechanisms to explain why specific homeotic genes are expressed in one body segment and not in a neighboring segment. By using genetic approaches, it was demonstrated that the spatial and temporal expression of the homeotic genes was enhanced by the Trithorax group of genes and repressed by the Polycomb group of genes in a segment-specific manner.1 However, the manner in which the Trithorax and Polycomb groups of proteins controlled homeotic gene expression was not initially understood. Investigators using yeast and Tetrahymena discovered that enzymes that modify histones (the packaging structure for DNA) could silence specific genes.2–4 Biochemistry work revealed that the histone-modifying enzymes that repress gene expression contain a SET domain that catalyzes histone methylation. The importance of histone methylation in Drosophila species development became apparent when the Polycomb protein complex was shown to have a SET domain–containing enzyme capable of methylating H3K9, a repressive epigenetic mark.5,6 Similarly, the Trithorax proteins TRX and Ash1 were found to contain SET domains and capable of methylating histones with a mark that facilitates active gene expression.7–9 Taken together, these studies reveal that histone modifications are well-conserved phenomena that are critical for regulating gene expression profiles during development. The creation of Dolly the sheep also revealed important features of epigenetic mechanisms. Wilmut et al.10 sought to clone a sheep by taking the nucleus from a differentiated cell (mammary gland cell) and placing that nucleus into an unfertilized oocyte (developing egg cell). Although they were eventually successful, this process of nuclear transfer was extremely inefficient. This work revealed that although the mammary gland cell DNA contains the blueprint necessary to develop an entire sheep, it was not easy to revert a differentiated nucleus back to a pluripotent state. It became apparent that although the blueprint of DNA is the same in an embryonic stem (ES) cell and a differentiated cell, nonmutation-based changes have occurred to the DNA in the differentiated cell that prevent the simple reversion or manipulation of a cell’s identity. This study supports the idea that, as cells differentiate, they accumulate stable and heritable changes in their nucleus that restrict gene expression patterns and define identity. These changes are, by definition, epigenetic. The study of epigenetic phenomena presupposes that genes can be marked as transcriptionally “on” or “off” by modifications that are independent of the primary nucleotide coding sequence. The DNA in each cell is composed of approximately 3 × 109 nucleotide bases that could presumably stretch to 2 m in length. To facilitate efficient packaging, DNA is wrapped in a threadlike fashion around protein spools called histones. The histone octamer consists of two molecules of each histone: H2A, H2B, H3, and H4.11–13 The histone octamer and the 146 bp of DNA looped around it forms a structure called a nucleosome, the functional unit of chromatin. In addition to serving as packaging facilitators, nucleosomes are amenable to modifications and dynamic remodeling, which makes them an active participant in many chromosomal processes including transcription, replication, DNA repair, kinetochore and centromere construction, and telomere maintenance. The regulation of gene expression at the chromatin level occurs by: (1) covalent histone tail modifications; (2) adenosine triphosphate (ATP)-dependent chromatin remodeling; and (3) DNA methylation (Figure 24-2).14 This chapter focuses on covalent histone tail modifications and ATP-dependent chromatin remodeling. The amino terminal tails of histones protrude and are subjected to various covalent posttranslational modifications (e.g., methylation, acetylation, sumoylation, phosphorylation).11 These histone tail modifications regulate whether the neighboring DNA is transcriptionally active (euchromatin) or repressed (heterochromatin). Activating histone modifications attract ATP-dependent nucleosome remodeling factors that can reposition or eject histones, thus promoting transcription or repression of the neighboring DNA. Thus, complex interplay between covalent histone modifications, ATP-dependent chromatin remodeling enzymes, and the transcriptional machinery (i.e., DNA binding factors, transcription initiation, elongation complexes) determines whether genes are actively expressed or repressed. Figure 24-2 A, DNA provides the template that encodes all potential genes. Not all genes are expressed in all cells. Chromatin provides a way of partitioning this genetic information into active and repressed components. The three mechanisms whereby chromatin can modify the expression or repression of genes includes DNA methylation (Me), chromatin remodelers (blue oval), and histone modifications (e.g., phosphorylation, methylation, acetylation) imparted by histone modifiers. (Adapted from Chang CP, Bruneau BG: Epigenetics and cardiovascular development. Annu Rev Physiol 74:41–68, 2012.) From an epigenetic standpoint, the two most important tail modifications are acetylation and methylation.15 Histone tail acetylation is associated with transcriptionally active chromatin. In contrast, histone tail methylation can be associated with either euchromatin or heterochromatin, depending upon the extent of methylation (mono- [me], di- [me2], or tri- [me3] methylation) and the specific tail residues that are modified. For example, lysine (K) residues available for methylation include K4, K9, K27, and K36 of histone H3, and K20 of histone H4. Methylation of H3K9 and H3K27 is associated with gene repression gene expression,16 whereas dimethyl (H3K4me2) and trimethyl (H3K4me3) marks at H3K4 are associated with actively expressed genes.17 As shown in Figure 24-3,18 the extent, specific lysine residue, and location of different histone methylation marks around a gene provide a signature for the transcriptional state of that gene. For example, H3K27me3 marks (black line) are located around the 5′ regulatory region of silenced genes. In contrast, H3K4me3 marks are highly enriched at the 5′ transcription start site of actively expressed genes. In contrast, H3K4me3 marks are not as enriched around poised and silenced genes. Figure 24-3 Epigenetic signatures differ around genes that are repressed (red rectangle), poised (black rectangle), and active (green rectangle). H3K27me3 marks (red line) are enriched at the 5′ region of silenced genes H3K4me3 marks (green line) are enriched at actively expressed genes. Thus active genes show enrichment of H3K4me3 marks and a paucity of H3K27me3 marks at the 5′ region. H3K36me3 (yellow line) marks are enriched at active genes. This illustrates the histone code hypothesis, which suggests that the epigenetic signature of individual genes, determines whether that gene is actively expressed or repressed. (Adapted from Lee BM, Mahadevan LC: Stability of histone modifications across mammalian genomes: implications for “epigenetic” marking. J Cell Biochem 108:22–34, 2009.)
Epigenetics in Cardiac Rhythm Diseases
Definition of Epigenetics
Historical Experiments That Illustrate the Salient Features of Epigenetics
Basis of Epigenetic Phenomena
Histone Tail Modifications
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thoracic Key
Fastest Thoracic Insight Engine
