Fig. 8.1.
CLSM 3D reconstruction of an A. fumigatus -infected lung. This picture has been taken from an infected and subsequently explanted murine lung by use of confocal laser scanning microscopy (CLSM). A 3D rendering was done based on the microscopic sample sections. The organ-specific structures such as alveoli were detected by their autofluorescence signal. The small red clouds are so-called neutrophil extracellular traps (NETs), which are composed of extracellular DNA fibers and were released during the neutrophilic immune reaction towards the fungal pathogen. NETs were stained with a DNA-specific dye
In order to provide a certain flexibility regarding the parallel detection of different fluorescence colors, common CLSM systems are equipped with several lasers and a combination of adjustable excitation and emission filter sets. Very recent machines have been released that use white lasers as light source and can very quickly and precisely tune acousto-optical beam splitters and filters on both the excitation as well as emission side. These systems allow the use of almost all fluorophores that possess a spectral excitation/emission peak in the visible light spectrum.
One disadvantage of note is the speed of image acquisition in a CLSM. As every single two-dimensional (2D) picture is based on the scanning process of the whole specimen in one plane, it takes a certain time until the required digital information for one image is obtained (in contrast to a widefield system in which the picture is recorded by the camera as soon as the specimen is illuminated by the transmission light). Although it is possible to analyze cellular behavior using CLSM (Gunzer et al. 2004), fast moving cells sometimes generate problems when their tracks are the question of the analysis. These problems become even bigger as soon as the cellular movements are analyzed in four dimensions (3D over time). However, novel developments such as resonant scanners and extremely fast and sensitive detectors have helped to partly overcome these problems. Today, the efficiency of light generation from the sample, rather than the slow detection speed of the CLSM, is the key factor limiting the recording speed of a given system.
C. Electron Microscopy
In 1873, Ernst Abbé published a landmark equation that easily correlates the wave nature of light or electrons and the lens properties, thereby calculating the resolving power of a given optical system.
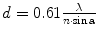 | d | Resolution |
λ | Wavelength of the light source | |
n | Refractive index of the medium | |
α | Aperture angle of the objective |
The value d defines the smallest distance between two objects such that they can still be detectable as two separate entities. Thus, the smaller the value for d, the better resolution a microscope has. The equation clearly shows that d is highly dependent on the wavelength λ, where short wavelengths are obviously desirable. For normal light microscopy, light in the range of the visible light spectrum (>380 nm) is usually used, resulting in a maximum resolution of around 200 nm. The wavelength of an electron beam is dramatically smaller. In modern electron microscopy (EM) systems, values around 2 pm are achievable, suggesting a theoretical resolution of ~1 pm. However, the practical resolution with these machines is estimated to be ~0.1 nm due to aberration errors of the electron lenses. This is still good enough to see individual atoms (Meyer et al. 2008).
Inside these microscopes, an electron beam is generated by the emission of electrons from a glowing cathode. These electrons are subsequently accelerated in a high vacuum by a tunable anode and aligned by a system of electronically charged lenses. In the field of electron microscopy, two principally different major technologies are known: transmission EM (TEM) and scanning EM (SEM) (Koning and Koster 2009).
In TEM, the ray of electrons is shone through an extremely thin sample of interest (Fig. 8.2a). Upon interaction with the atoms in the sample, the electrons are scattered and/or absorbed. A detection device behind the sample is then able to generate a magnified image of the modified electron beam that can be observed on a screen or with a camera. TEM thus generates highly resolved images of thin sections of samples. In contrast, SEM uses an electron beam that is scanned over the surface of a fully intact 3D specimen and releases secondary electrons from its surface. These are measured by a detector and generate one pixel of a digital image. By scanning over the surface of the sample very detailed 3D images of surfaces can be obtained (Fig. 8.2b). We have used this approach to document the details of phagocyte contacts to A. fumigatus spores (Behnsen et al. 2007a) as well as the fine structures of DNA NETs with these fungal elements (Bruns et al. 2010). Others have used SEM and TEM to document the uptake of Aspergillus spores by alveolar macrophages (Ibrahim-Granet et al. 2003).
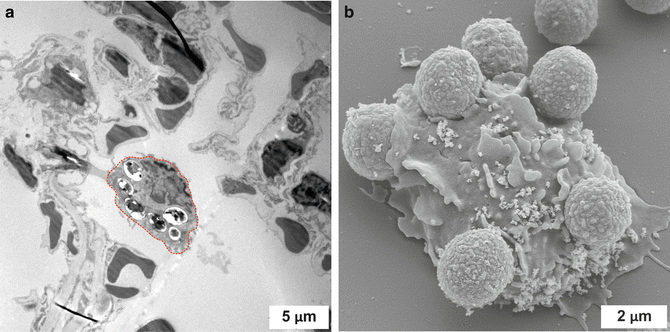
Fig. 8.2.
Different appearance of TEM and SEM images. (a) Transmission electron microscopy (TEM) image of the situation in an A. fumigatus-infected mouse lung. An alveolar macrophage (outlined in red) scans the alveolar lumen and has already taken up six fungal spores. (b) The scanning electron micrograph (SEM) visualizes the in vitro co-incubation of isolated murine neutrophils and spores of the mold A. fumigatus after 4 h. It is nicely observable that the immune cell has started to internalize some of the attached fungal particles. Scale bars are depicted in white. The pictures were taken in collaboration with Prof. Manfred Rohde at the Helmholtz Center for Infectious Diseases in Braunschweig, Germany
Although TEM images give an extremely good resolution of cellular structures, they are typically only 2D. A fairly recent addition to the technology now allows the generation of 3D reconstructions from TEM images without the need for serial sections. This technique is called electron tomography (ET). Here, the thin sample is placed on a rotating device and a series of TEM images from different angles is taken, which then allows reconstruction of the 3D network of observed structures with unprecedented detail (Koning and Koster 2009). This technology has already been used to study the ultrastructure of fungal elements (Hohmann-Marriott et al. 2006). It would be extremely interesting to also use ET for the study of host–pathogen interactions.
Thus, while the advantage of EM obviously lies in its extreme resolving power, the downside is its inability to image live specimens. Furthermore, EM requires extensive sample preparation and a lot of experience both in the use of the microscopes as well as in interpretation of the data. Nevertheless, without doubt the electron microscope has opened to human eyes the fascinating ultrastructural world, which has always intrigued scientists and laymen and will continue to do so in the future. The inability of EM to image live specimens is partly overcome, albeit not with the same resolving power, with new methods in optical microscopy (see next section).
D. Super-Resolution Light Microscopy
For more than 100 years the obtainable maximum resolution of optical microscopes was accepted to be ~200 nm, based on Abbés rules of diffraction (Hell 2009) (see also the previous section on electron microscopy). However, since the 1990s a number of innovative advances have been made that allow resolutions of up to ten times higher using currently available commercial light microscopy systems. Here, we will discuss three key developments in the field: stimulated emission depletion, structured illumination microscopy, and localization microscopy. Unfortunately, not much use has been made of these developments in experimental mycology so far. For a fine review of the use of these novel systems in the general microbiology field the reader is referred to (Coltharp and Xiao 2012). Future work should thus aim at exploiting these techniques for investigating fungal infections and the interaction of fungi with the vertebrate immune system in unprecedented detail.
STED (stimulated emission depletion): STED is principally an advanced method of CLSM. However, rather than using just one blue exciting laser spot, the dimensions of which are limited to the 200 nm size defined by Abbé optics, STED projects a second, red-colored, doughnut shaped depletion beam over the exciting beam. This depletion beam has a hole in the middle, where no laser light is emitted. All fluorescence hit by the depletion laser is immediately quenched again due to a process called stimulated depletion. The key feature of STED is that the hole in the center of the doughnut can be adjusted to be significantly smaller than 200 nm, based on the power and the optical shaping of the laser. Thus, the combination of excitation spot and depletion doughnut allows the generation of a much smaller spot of residual excitation than could be obtained with a focused excitation laser alone (Eggeling et al. 2009). This small spot of residual excitation increases the optical resolution in the x,y -plane by a factor of four to eight. In modern commercial systems, this is indeed achieved by the click of a mouse on the control software. The advantage in resolution is bought at the expense of the very bright light of the depletion beam. Thus, STED imaging is prone to rapid bleaching and phototoxicity, although novel developments such as gated STED reduce this problem (Vicidomini et al. 2011). Furthermore, not all dyes are suitable for STED imaging and there is only the possibility to simultaneously image two colors as opposed to other multicolor options. Finally, STED only increases the resolution in the x,y-plane, while resolution in the z-direction is not changed.
SIM (structured illumination microscopy): SIM uses the projection of a known grating structure into the imaging plane that is recorded together with the emitted fluorescence. By interference with the emitted fluorescence, so-called moiré fringes are generated. These contain additional structural information about the sample that cannot be simply extracted by optical resolution but needs complex mathematical operations (Fourier transformations) to obtain an image. As a result, SIM increases the resolution in all three directions of space by a factor of at least two (Schermelleh et al. 2008; Gustafsson 2005). Thus, although not as good as STED, the principal advantage of SIM is the fact that it works with any fluorescent dye and does not use high intensities of light. Thus, it can also be used for long-term time-lapse imaging (Fiolka et al. 2012).
Localization microscopy (PALM/STORM): These approaches exploit the ability of fluorescent dyes to emit light only for short amounts of time, whereby two adjacent molecules rarely emit simultaneously. However, this is typically not seen because conventional fluorescent images are an integrated fluorescence signal of many molecules permanently illuminated over a long time. But, when a given field of fluorescent molecules is excited with stroboscopic light rather than continuous illumination, at one stroboscopic pulse only a fraction of molecules will fluoresce. When the next stroboscopic pulse hits the system, a separate fraction of molecules will glow. If, by this approach, a large series (typically several thousand images) of stroboscopic images is taken, for statistical reasons all available fluorescent molecules will have glowed at least once. The important feature is that by this approach two directly adjacent molecules will not shine on the same frame of the series, but only on separate frames. This fact allows their optical separation as two molecules, rather than as one larger fluorescent entity. The image generated by this method is not a classical picture like that made by a PMT. Instead, from each light spot detected in the series of images the exact center of the generating fluorescent spot will be calculated from fitting the Gaussian light distribution with nanometer precision. This calculated molecular position will be artificially set as the position of a positive pixel whose superimposition with all other found spot positions generates the final super-resolution image. As a result, localization images have a “pointillist” appearance but their resolution (or rather, their precision in exactly localizing the source of a fluorescence signal) in the x,y -plane can reach values of 20 nm or better (Coltharp and Xiao 2012; Hell 2007). To generate this extreme resolution, PALM/STORM requires very specific dyes and experimental conditions (such as stringent buffer conditions on fixed samples). This renders the technology rather complicated and limited in its use. Nevertheless, commercial systems are available and if the requests by researchers rise, it is expected that suppliers will respond with developments towards greater ease of use.
E. Flow Cytometry
A very powerful approach for analyzing host–fungal interactions is flow cytometry. Here, rather than imaging a few cells or fungal elements with very high resolution, many cells (typically many thousands) are imaged in a short time with only limited resolution. Flow cytometry uses a constant flow of liquid droplets to enclose individual cells or cell pairs. These cell-containing droplets subsequently cross one or more laser beams. By hitting intra-and extracellular structures, the laser light is diffracted and scattered light is produced. Light that is diffracted on the surface of the spherical particles is then detected by a spectral detector in a 180° angle relative to the excitation beam (forward scatter, FSC). The diffraction angle allows the calculation of the cell size so that this detector (i) counts all cells that pass the detection optics and (ii) assesses their individual relative size. A second detector is located (side scatter, SSC) at a 90° angle. The amount of light detected here directly correlates with the amount of intracellular structures (granules) of a cell or cell–cell pair.
In addition, cells can be labeled with a variety of fluorescent agents that bind to their surface or intracellularly. If, for example, a fluorescently labeled monoclonal antibody is used that can bind to a specific surface structure of a cell, this cell type can be selectively labeled in a heterogeneous cell suspension (Fig. 8.3). By running many thousands of cells through the flow cytometer and counting all labeled cells, the relative number of these cells in a mixture can be easily obtained. Introduced as principal technology in 1969 (Hulett et al. 1969), flow cytometry has revolutionized many scientific fields, especially immunology. Importantly, in addition to imaging cells, flow cytometry can also be used to physically sort them according to predetermined parameters of scatter and fluorescence (fluorescence activated cell sorting, FACS). There are many applications in the field of antifungal immunology that can benefit from the technique, e.g., measuring the genomic response of sorted epithelial cells after phagocytosis of Aspergillus spores (Gomez et al. 2010) or quantifying the organ-specific immune response after invasive candidiasis (Lionakis et al. 2011), the response of mast cells towards the fungal cell wall component zymosan (Yang and Marshall 2009), or the uptake and killing of conidia by diverse phagocytyes (Jhingran et al. 2012).
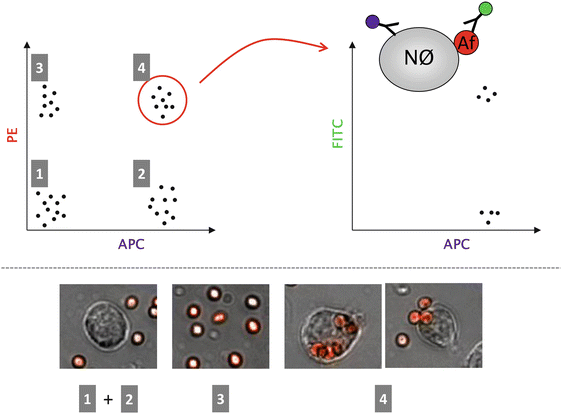
Fig. 8.3.
Quantification of phagocytic events by flow cytometry. Example of how phagocytic events can be analyzed by a flow cytometric approach. The background of this experiment were mice that had been infected intratracheally with genetically modified fungal spores, expressing the red fluorescent protein tdTomato. After an incubation time of several hours, the immunological infiltrate was washed out of the lung by a bronchoalveolar lavage followed by monoclonal antibody staining specific for neutrophil granulocytes (clone 1A8 coupled to the fluorescent dye allophycocyanine, APC). After flow cytometric analysis, four clearly distinguishable cell populations are visible by plotting the APC fluorescence versus the tdTomato signal detected in the PE channel (see left part of Fig. 8.1): Population 1 constitutes cells that are not neutrophils and that are not co-localized with any fungal spores. Cell population 2 consists of neutrophils that are also not in contact with the fungal particles. The majority of population 3 is most likely composed of free fungal spores but also other (immune) cells that are in contact with the spores are found here. Cell population 4 is the group of interest, where many neutrophils are found that have internalized at least one or more spores. These cells are double-positive for both APC as well as the tdTomato signal. However, especially for neutrophils, it is well known that these cells, besides phagocytosing fungal morphotypes, also attach these entities to their surface, thereby collecting a large number of them. This complex is of course also double-positive for both types of fluorescence and hence generates a potential source of error for this quantitative approach. One way to eliminate false positive events is to add a second antibody (labeled with the green fluorophore fluorescein isothiocyanate, FITC) specific for the fungal spores (AF). Neutrophils (Nϕ) that have spores attached to their surface are then additionally stained green so that these events can be excluded from the analysis. Please note that the image only shows theoretical flow cytometry data for reasons of simplicity. Real dot plots contain many hundreds to thousands of dots
In general, any approach that allows the fluorescent labeling of fungal elements and immune cells separately, in vitro, within true infection models, or even from patient samples, is able to be analyzed on a large scale by flow cytometry.
A critical limitation of flow cytometry is that it is not able to show individual events directly. Instead they are transformed into dot plots or histograms as output graphs. But, if for example, phagocytosis is the investigated process, flow cytometry is hardly able to distinguish whether co-associated fungal elements and immune cells are only attached to each other’s surfaces or whether a phagocyte has entirely internalized the pathogen. A novel invention called imaging flow cytometry has changed this. Here, in principle, the same hardware setup as inside a flow cytometer is used. However, rather than just recording the total fluorescence of an event passing the laser beams, it is photographed by a fast, multicolor camera (Barteneva et al. 2012). At first glance the resulting data look like conventional flow cytometry plots but they allow a detailed look at each single dot of a dot plot as a photographic image. Here, phagocytosis can easily be distinguished from simple attachment. This technology has been used to show that treatment of mice with the drug sulfasalazine during pulmonary pneumocystosis enhances fungal uptake by alveolar macrophages and leads to more rapid clearing of the fungal infection (Wang et al. 2010). Thus flow cytometry, with its ability to rapidly scan and/or sort cells in large numbers, and imaging flow cytometry are two very potent imaging technologies, both in experimental as well as clinical mycology.
However, although important observations can be made in a reduced environmental complexity in vitro, one has to keep in mind that an in vitro situation never fully represents the in vivo environment of a living organism. Therefore, it is always important to think about experimental approaches that allow the in vivo observation of the investigated phenomena. This is more complicated to establish and often limited in its application possibilities but the results obtained from intravital imaging experiments deliver more meaningful results. In fact, due to the increasing availability of in vivo imaging techniques, observations made purely in vitro are nowadays often considered as potentially suffering from artifact. As a result, researchers wishing to publish work on dynamic cellular processes today are often asked to verify their in vitro data in an in vivo setup. To help find the right approach and judge the feasibility of a reviewer’s comment,we will now discuss different systems that can generate intravital imaging data.
F. Intravital 2-Photon Microscopy
The principal construction of an intravital 2-photon (2-P) microscope is similar to the CLSM, as discussed above. Thus, a laser beam is scanned over the sample and the generated fluorescence is detected by PMTs and reconstructed into a full multicolor picture (Niesner et al. 2008a; Niesner et al. 2007). However, the difference lies in the use of the laser and the physics behind the generation of the fluorescent signal. For the generation of fluorescence, a fluorophore needs to absorb an incoming photon that has the correct wavelength so that the delocalized electrons of the fluorophore can be lifted from a ground state to an excited state. Upon return to the ground state, the electrons emit a fluorescence photon that is slightly red-shifted in relation to the exciting photon. Hence, the color of fluorescence is slightly redder than the color of excitation. Typical fluorophores need excitation wavelengths in the visible spectrum. However, the principal problem of visible light photons is that they cannot penetrate very deeply into thick tissue due to massive diffraction. As a result, intravital microscopy with visible light using a conventional CLSM is possible, but does not reach large depths in tissue (Gunzer et al. 2004; Stoll et al. 2002). Importantly, wavelengths between 800 and 1,200 nm (near infrared) have excellent tissue penetration capabilities and thus would be perfect for imaging in vivo (Konig 2000). However, conventional fluorophores cannot be excited by these photons because they possess too little energy.
This is where the 2-photon process comes into play. If laser radiation of near-infrared color is very intense, one fluorophore can be hit at almost the same time by two or more of these red photons. Then, they combine their energy on the surface of the fluorophore and excite it to emit normal fluorescence. At the same time, this allows the use of the red laser wavelengths that give good tissue penetration. The enormous intensity of laser light required for 2-PM is generated by the use of pulsed rather than continuous wave lasers (as in conventional CLSM). Pulsed lasers (typically titanium–sapphire solid-state lasers) have immense amounts of photons in extremely short-lived pulses (10−13 s pulse duration). When these are highly focused by the optics of the 2-PM system, the energy at the focal spot is high enough to allow the 2-P process to occur at a meaningful frequency that enables imaging (Germain et al. 2006). At the same time, the system is inherently confocal as no out-of-focus fluorescence is generated because the extreme level of photon flux required for the excitation of fluorescence is only reached at the center of the focal spot of the optics.
2-PM is the most widely used technology for high-resolution intravital imaging in experimental animals and we have made extensive use of the technology to investigate antifungal immunity (Hasenberg et al. 2011c; Nitschke et al. 2008; Bruns et al. 2010). But, 2-PM offers more than just the ability to image fluorescent structures deep inside scattering tissue. One powerful approach is fluorescence lifetime imaging (FLIM). For FLIM, not only the intensity of fluorescence as such, but also its decay characteristics after a single excitation are measured. This can be nicely employed to study the characteristics of endogenous fluorescent molecules such as NAD(P)H. This important coenzyme has a characteristic 2-P excitation at 760 nm and an emission peak at around 460 nm. Within a cell (e.g. a neutrophil granulocyte), NAD(P)H can exist as the free coenzyme. This has a fluorescence lifetime of ~430 ps. However, there are more than 100 enzymes known that can bind NAD(P)H as a coenzyme, and when bound to a protein the lifetime of NAD(P)H changes quite substantially. We have shown that this feature is able to very specifically detect the NAD(P)H molecules inside of a neutrophil granulocyte because they are bound to NAD(P)H oxidase, the key enzyme for the generation of fungitoxic reactive oxygen species (Niesner et al. 2008b). When bound to the oxidase, the lifetime of NAD(P)H was stretched to 3.6 ns. Spots with this lifetime showed a characteristic enrichment at sites where neutrophils touched fungal elements and could be selectively quenched with the addition of inhibitors of the oxidase (Niesner et al. 2008b). Because there are many more enzymes to which NAD(P)H can bind, it is tempting to speculate that each one shows a characteristic lifetime of the associated NAD(P)H molecule and thus can be identified by FLIM without the need for additional staining. In addition, other endogenous fluorophores (e.g., FAD) exist in cells and are able to be investigated by FLIM. Thus, together with the ability for intravital imaging, 2-PM and FLIM are powerful tools for the investigation of host–fungal interactions in vivo.
G. Whole-Body Imaging
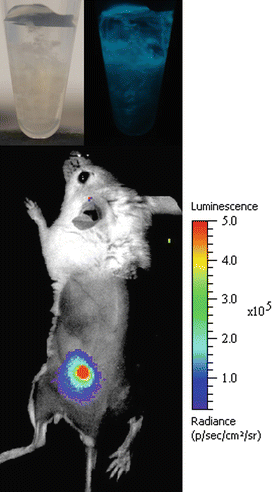
Although being very potent for high-resolution imaging at organ level, 2-PM is also limited in its penetration/detection depth. Depending on the type of tissue, several hundred micrometers can be achieved, but it is not possible to scan through larger organs or even whole intact animals. Thus, it is not possible to use the 2-PM technique for whole-body imaging, e.g., if the position or infectious spread of a fungus is to be monitored inside of an experimental animal. To close this gap, different companies offer so-called whole-body imaging systems for small animal research. The first machines that entered the market used the detection of bioluminescence signals with very sensitive CCD cameras as their key function. Bioluminescence makes use of luciferases, which are enzymes made by insects, corals, or copepods. By the breakdown of their substrates (e.g., luciferin or coelentaerazine), luciferase is able to generate photons that can be detected with sensitive cameras, even if they are generated deep inside the tissue. The big advantage of this enzyme system lies in the spontaneous release of photons during the chemical reaction without an exciting light source that also would have to pass through the surrounding tissue. Such bioluminescent reporter systems allow for real-time imaging and longitudinal infection studies, thereby reducing the number of monitored animals and decreasing experimental variability. However, in contrast to bacterial bioluminescent reporter systems, eukaryotic systems require the addition of an extrinsic substrate. Accordingly, substrate stability, tissue penetration, and uptake by the pathogen are limiting factors for bioluminescence imaging of fungal infections. Based on pioneering studies in the human commensal Candida albicans (Doyle et al. 2006; Enjalbert et al. 2009), we have expressed the Gaussia princeps copepod luciferase on the surface of A. fumigatus to demonstrate that cutaneous aspergillosis is clearly detectable in infected animals (Fig. 8.4) and dependent on the innate immune system for growth control (Donat et al. 2012). Others have shown that pulmonary and systemic infections with A. fumigatus can also be monitored by this method when expressing the firefly luciferase from Photinus pyralis intracellularly (Brock et al. 2008; Jouvion et al. 2012; Ibrahim-Granet et al. 2010). Meanwhile, such whole-body scanners also make use of the detection of fluorescence signals. With the introduction of a variety of red and far-red dyes it became possible to excite these fluorophores from the outside with light of a very long wavelength. This relatively energy-poor light is able to penetrate biological tissue very efficiently, thereby enabling the use of fluorescent probes for this application. Bioluminescent fungal strains interacting in vivo with fluorescently labeled immune cells may therefore provide a deeper understanding of the intimate host–pathogen interplay during infection.