CHAPTER 97 Ventricular Assist Devices
Left ventricular assist devices (VADs) have become the standard of care for patients with refractory, end-stage heart failure.1,2 Several studies have demonstrated the superiority of mechanical support over optimal medical therapy with respect to improving survival as well as quality of life in this subgroup of patients.3–5 Over the past decade, there has been a significant evolution in VAD-related technologies. Devices have undergone substantial modifications in an effort to improve survival while on support, increase durability, and limit device-related infections, device thrombosis, device malfunction, and perioperative bleeding.6,7 Improvements in device design have also yielded devices that can be implanted as destination therapy.
Current devices can be put into subcategories on the basis of their features: short-term versus long-term devices, paracorporeal versus intracorporeal devices, pulsatile versus continuous-flow devices, full versus partial support devices, and assist devices versus complete heart replacement (i.e., total artificial heart). The indications for implantation of a mechanical device have expanded over time and include acute cardiogenic shock, bridge to transplant (BTT), bridge to decision, and destination therapy (DT).8,9 This chapter will present a complete overview of VADs, including a historical perspective, indications for mechanical support, important factors related to patient selection, types of devices, results using mechanical support, surgical technique, postoperative management, and complications that occur with mechanical support.
HISTORICAL PERSPECTIVE
A major victory for mechanical support came in 2001 with the results of the REMATCH trial.3 This prospective, randomized trial of patients with end-stage heart failure who were not eligible for transplantation demonstrated a significant survival advantage for patients who underwent implantation of a mechanical assist device as compared with patients who were managed with optimal medical therapy. The study, however, also highlighted various complications of mechanical support, such as bleeding, infection, stroke, device malfunction, and development of right heart failure, which became a primary focus for device companies and engineers in subsequent years.
INDICATIONS FOR MECHANICAL SUPPORT
Indications for mechanical support include acute cardiogenic shock after acute myocardial infarction (AMI), postcardiotomy cardiogenic shock, myocarditis, refractory ventricular arrhythmias, chronic heart failure as a bridge to transplant, and chronic heart failure destination therapy.10–15
Cardiogenic Shock after Acute Myocardial Infarction
Approximately 6% to 7% of all patients with a myocardial infarction develop cardiogenic shock as a complication of the MI, and the vast majority of these are ST-elevation (Q-wave) MIs (STEMI).16 When cardiogenic shock complicates an acute MI, the reported associated mortality has traditionally been 85% to 90%.17 For patients with refractory cardiogenic shock despite maximal pressors, inotropic support, and an intra-aortic balloon pump (IABP), mortality approaches 100%.18 Implantation of a mechanical assist device to provide complete circulatory support may be the only viable option for survival in these cases.19,20,21 By providing complete circulatory support, mechanical circulatory assistance can reverse hypotension, maintain vital organ perfusion, and maintain adequate coronary perfusion pressures.22
The preferred options for short-term mechanical ventricular support after an AMI include the CentriMag (Levitronix LLC, Waltham, MA) ventricular support system, the Abiomed (Abiomed, Inc., Danvers, MA) BVS 5000, and the Abiomed AB5000.23,24 Each of these devices is designed for short-term use. The goal of mechanical support in patients with cardiogenic shock after an AMI is to bridge the patient to a second procedure, which includes percutaneous coronary intervention, coronary artery bypass grafting (CABG), CABG plus valve, implantation of a long-term LVAD, and explantation of the short-term LVAD or right VAD (RVAD), or both, after myocardial recovery.
The CentriMag consists of an extracorporeal centrifugal pump that combines the drive, magnetic bearing, and rotor function into a single unit.23,25 It has several key advantages over the Abiomed BVS 5000 and AB5000. The device is relatively easy to assemble, prime, and implant. All three devices can produce flows of up to 10 L/min and can provide isolated left ventricular support as an LVAD, isolated right ventricular support as an RVAD, or biventricular support as a full-support biventricular assist device (BiVAD).23,25–27 Placement of a BiVAD allows decompression of both the left and the right ventricle, restores hemodynamic stability, provides enhanced peripheral perfusion, and prevents end-organ dysfunction. Advantages of the Abiomed AB5000 over the CentriMag include that it allows patients to ambulate and that it is approved for medium-term support, as opposed to the CentriMag, which is approved for only 6 hours of support.28
Cardiogenic Shock after Cardiotomy
Patients in cardiogenic shock after cardiotomy are also best treated with a short-term VAD, such as the CentriMag, Abiomed BVS 5000, or Abiomed AB5000.29–31 Another advantage of the CentriMag VAD in this subgroup of patients is that the same cannulas used for cardiopulmonary bypass (CPB) for the cardiac surgery operation can be used for the VAD circuit, obviating the need for creating an anastomosis, which is required with the Abiomed devices.23,25 Like the goal of mechanical support in patients after an AMI, the goal of mechanical support in patients with cardiogenic shock after cardiotomy irrespective of which mechanical device is used, is to bridge the patient to a second procedure, which includes implantation of a long-term LVAD, as well as explantation of the short-term LVAD after myocardial recovery.29–31
Myocarditis
Acute heart failure and cardiogenic shock from myocarditis are typically seen in a younger patient population than the other subgroups. Because the probability of recovery is relatively high in these patients, they should receive a short-term device, such as the CentriMag, Abiomed BVS 5000, and Abiomed AB5000.32 Generally, our preference is to implant a biventricular device for biventricular support in these patients. After normalization in end-organ perfusion and function, myocardial function and recovery are assessed, according to our weaning protocol (see Patients in Cardiogenic Shock after Acute MI or Cardiotomy, later).
Refractory Ventricular Arrhythmias
The subgroup of patients with refractory arrhythmias represents a very heterogeneous group of patients. Some do not have significantly reduced cardiac function, whereas others have severely depressed ejection fractions.8,9,12 They have all failed pharmacologic therapy, such as amiodarone, lidocaine, and beta-blockers, to control their arrhythmias. They have all undergone insertion of a defibrillator, which has also failed. Among the patients with reduced left ventricular function, some have preserved right ventricular function, but in others, function of both the left and right ventricles is reduced. Our preference is to implant an isolated LVAD, which is generally successful in eliminating the arrhythmias, unless the right ventricular function is severely reduced as well, in which case we implant a long-term BiVAD, generally an intracorporeal BiVAD.
Chronic Heart Failure as a Bridge to Transplant
Progression of chronic heart failure is characteristic of the largest subgroup of patients who receive LVADs.33,34 These patients are typically already on a heart transplant list. Although most of these patients chronically deteriorate to the point that they require mechanical support, a subset will have an acute exacerbation of chronic heart failure because of a new acute infection, an arrhythmia, or a new area of myocardial ischemia. These patients typically do well after placement of a long-term VAD, such as the HeartMate XVE, HeartMate II, or Thoratec VAD.1,2,35–37 They are often discharged from the hospital after their VAD is placed and return at a later date for a transplant.4,5
Chronic Heart Failure as Destination Therapy
Patients with end-stage heart failure who are not candidates for heart transplantation might be considered for permanent implantation of an LVAD, or destination therapy (DT). It is of paramount importance to implant these patients with devices before the onset of multisystem organ dysfunction or failure, and before they become nutritionally and metabolically compromised. Mortality increases to an almost prohibitive value with the onset of end-organ failure, such as renal failure, and in the presence of active infection. In a study of 280 patients who underwent HeartMate XVE LVAD implantation as DT in the post-REMATCH era, the most important determinants of in-hospital mortality were poor nutrition, hematologic abnormalities, markers of end-organ or right ventricular dysfunction, and lack of inotropic support.38 Currently, the only FDA-approved device for DT is the HeartMate XVE. Several newer, miniaturized pumps, including the Thoratec HeartMate II (HM II), the VentraCor VentrAssist, and the Terumo DuraHeart are currently being evaluated or are scheduled to be evaluated for DT.
PATIENT SELECTION
Cardiac Factors
Prudent patient selection is a critical element in achieving good clinical results with LVADs. Numerous studies have identified independent predictors of adverse outcome after LVAD implantation.39–41 The common denominator among these independent predictors of mortality is end-organ failure, such as respiratory failure requiring mechanical ventilation, renal failure requiring dialysis, shock liver with markedly elevated transaminases, and diffuse coagulopathy resulting from malfunctioning clotting mechanisms. It is important to implant mechanical support in patients before end-organ failure develops. This applies to patients with chronic heart failure with an acute decompensation who are receiving long-term VADs, such as the HeartMate XVE.40,41 For patients with acute cardiogenic shock without a history of heart failure, such as a patient with cardiogenic shock after an acute MI or a patient with acute myocarditis, who is receiving a short-term VAD for acute stabilization and a “bridge to decision,” end-organ failure is often present at the time of initial presentation and evaluation for a mechanical device.
It is important to identify those patients with significant right ventricular (RV) dysfunction before LVAD implantation, as RV failure after LVAD implantation could be a fatal complication.42,43 Preoperative predictive factors for RV failure after LVAD implantation include low preoperative mean pulmonary artery pressure and low RV stroke work index (RVSWI), which can be calculated by the following equation:
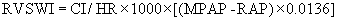
where CI is cardiac index, MPAP is mean pulmonary artery pressure, and RAP is right atrial pressure.43 Normal RVSWI is 8 to 10. A lower RVSWI implies increased chance for RV failure after LVAD implantation.43 These patients should be considered for BiVAD implantation instead of an isolated LVAD.
Patients with severe tricuspid regurgitation, or an atrial septal defect (ASD), or a patent foramen ovale (PFO) should have these lesions corrected at the time of VAD implantation using standard techniques. If an ASD or PFO is not corrected, it may result in severe hypoxia due to right-to-left shunting after the left ventricle is unloaded. Patients with severe aortic insufficiency should have their native aortic valve repaired or oversewn at the time of VAD implantation. For patients who are perceived to be candidates for being weaned to recovery, the aortic valve should be repaired by approximating the raphe of each leaflet.44,45 If the valve cannot be repaired, a tissue valve is preferred because it has a lower risk of thromboembolism than a mechanical valve. Any patient with a mechanical valve undergoing LVAD implantation should have the aortic valve oversewn with a patch or replaced with a tissue valve to prevent thromboembolism. Severe mitral stenosis needs to be repaired at the time of VAD implantation if it significantly interferes with inflow to the device.
Noncardiac Factors
Contraindications for device implantation include irreversible end-organ failure, particularly renal, hepatic, and respiratory, which are uniformly independent predictors of poor outcome.39–41,46,47 Severe, unrecoverable neurologic injury is also a contraindication for device implantation. Systemic sepsis poses a significant risk to patients undergoing LVAD implantation because it can cause a profound refractory vasodilatory state or lead to an increased incidence of device-related infections, such as device endocarditis.48,49
Preoperative renal failure is a significant independent predictor of mortality after VAD implantation.40,41,46 Thus, we try to avoid placing VADs in patients with renal failure requiring dialysis. This is particularly true for patients with acute exacerbation of chronic heart failure. Renal failure constitutes less of a contraindication for a patient presenting with acute cardiogenic shock without a history of heart failure, such as after AMI or from acute myocarditis. Several indicators of poor renal function that have been identified as predictors of outcome include urine output less than 0.5 mL/kg/hr despite diuretics, and a creatinine level greater than 5 mg/dL.
Active infection represents a relative contraindication for LVAD implantation.48,49 Ideally, patients should have two negative blood cultures over a 1-week period before LVAD implantation, because this demonstrates that the bloodstream is cleared of the infection. When time is limited, such as when a patient continues to decline hemodynamically despite maximal pressors and inotropic support, the team may be forced to proceed with LVAD implantation without the 1-week period of negative cultures. We recommend active involvement of an infectious disease physician for these cases.
Poor preoperative hepatic function has also been demonstrated to be an independent predictor of mortality after LVAD implantation.39–41 A prothrombin time of greater than 16 seconds is indicative of significantly decreased hepatic synthetic function and increases the chances for developing a diffuse coagulopathy intraoperatively and postoperatively. Diffuse coagulopathy can cause right heart failure secondary to increased transfusion requirements of blood and blood products, such as platelets, fresh-frozen plasma (FFP), cryoprecipitate, and factor VII.42,43 Development of right heart failure after LVAD implantation increases mortality.50 Another marker of hepatic function that has been correlated with survival after LVAD implantation is the preoperative bilirubin level.39–41
Reoperation also increases mortality,39–41 because it involves increased technical challenges, such as a reentry injury, increased CPB time, increased transfusion requirements, coagulopathy, and right heart failure. Another relative contraindication to LVAD implantation is the presence of a malignancy that entails a life expectancy of 2 years or less. Every patient requires individual attention and evaluation for appropriate decision making. A patient infected with the human immunodeficiency virus who is compliant with medical therapy, has a normal CD4 count, and has an undetectable viral load should be considered for an LVAD.39–41
In 2003, we published a revised screening scale to stratify the perioperative risk of VAD implantation.41 The screening scale was a result of our experience with 130 patients who underwent implantation of a HeartMate XVE LVAD. The following clinical factors were identified as preoperative independent predictors of adverse outcome: mechanical ventilation, prior cardiotomy, prior LVAD implantation, central venous pressure (CVP) greater than 16 mm Hg, and a prothrombin time of greater than 16 seconds.41 Each factor was given a statistically weighted score, with a score of greater than 5 being associated with a 46% mortality and a score of 5 or less being associated with a 12% mortality. It is important to note that this screening system is in the process of being updated, because it may not be applicable to patients undergoing implantation of an HM II device. Additionally, scoring systems that predict risk have limitations with respect to their positive and negative predictive values and should not be used as isolated instruments to make decisions about whether or not a patient should undergo LVAD implantation.
Deng and colleagues reported independent predictive factors for adverse outcome from a subset of 464 patients who underwent LVAD implantation.39 They concluded that sepsis, respiratory failure, right heart failure, age greater than 65, postcardiotomy shock, and prior AMI were risk factors for mortality.39
TYPES OF DEVICES
Devices for Short-Term Mechanical Support
Intra-aortic Balloon Pump
Insertion of an IABP was first reported by Kantrowitz in 1968. It has since become one of the most common forms of mechanical support for the failing heart. It is commonly used for high-risk percutaneous coronary intervention (PCI), post-MI acute cardiogenic shock, complications after MI, such as a VSD and ruptured papillary muscle, cardiotomy shock, and preoperative optimization of a patient who has had an MI and is awaiting CABG.51 Its mechanism of action involves inflation of the balloon in the descending aorta during diastole, which reduces afterload, increases coronary artery perfusion and pressure, improves myocardial oxygen supply, and decreases the myocardial oxygen demand.52 Counterpulsation can be synchronized with either the electrocardiogram or the arterial waveform. An IABP is usually placed under echocardiographic or fluoroscopic guidance, confirming the tip of the balloon to be located just distal to the left subclavian artery. Anticoagulation with systemic heparin is recommended to prevent thrombosis formation at both the insertion and the balloon sites. Vascular complications of the IABP include femoral artery rupture, pseudoaneurysm, and descending aortic dissection, as well as distal ischemia as a result of impedance of forward flow.53
Extracorporeal Membrane Oxygenation
For patients in circulatory as well as respiratory failure, extracorporeal membrane oxygenation (ECMO) may be a viable option (Fig. 97-1). This system employs a centrifugal pump with an oxygenator in the circuit, yielding complete CPB. The most commonly used ECMO pumps are the Biomedicus Bio-Pump (Medtronic, Inc., Minneapolis, MN) and the Sarns/3M centrifugal system (Sarns/3M, Ann Arbor, MI).54 Most survival data reported with ECMO have been in the pediatric literature, although some reports have described the experience in adults.55 We have limited our usage of the Biomedicus and Sarns pumps, because the anticoagulation requirements are high and durability is limited. We instead prefer to implant a CentriMag BiVAD as the pump for ECMO and to place an oxygenator into the RVAD circuit.
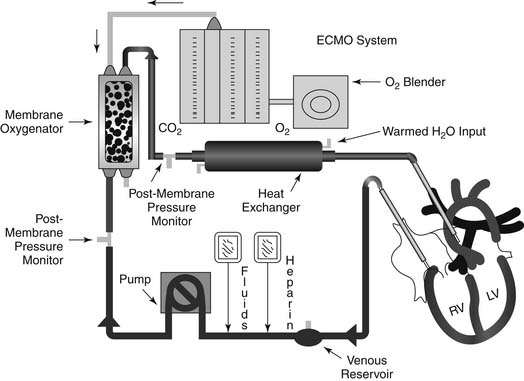
Figure 97–1 Extracorporeal membrane oxygenation circuit.
(Courtesy of EMedicine.com, used with permission.)
Bartlett recently reported a survival of 40% in 1600 patients undergoing ECMO for cardiogenic shock as part of the Extracorporeal Life Support Organization registry.54 Most of these patients, however, were pediatric patients. Series that have reported results primarily on adult patients have reported lower survival rates.
Levitronix CentriMag
The Levitronix CentriMag blood pump is an extracorporeal centrifugal pump that is FDA approved for up to 6 hours of support time and operates without mechanical bearings or seals (Fig. 97-2).23,25 The system combines the drive, magnetic bearing, and the rotor function into a single unit. The rotor is magnetically levitated, which enables the device to rotate without friction or wear, and eliminates heat production. This serves to minimize trauma to the blood and avoids mechanical failure. Because the rotor surface is uniformly washed, blood stagnation and turbulence in the pump are minimized. Hemolysis is reduced because the mechanical gaps in the pump are wider than 0.6 mm, allowing shear forces to be low. The device can produce flows of up to 10 L/min with a priming volume of 31 mL.23,25

Abiomed BVS 5000 and AB5000
The Abiomed BVS 5000 is a dual-chambered pneumatically driven extracorporeal pump that can be used for short-term support as a univentricular or biventricular assist device (Fig. 97-3).24 The dual-chambered polyurethane sacs fill with blood passively, and then pneumatically driven ejection occurs. The device is capable of flowing up to 6 L/min. Although in 1992 it was initially approved by the FDA for postcardiotomy shock, the indications for its implantation have now expanded to include post-MI cardiogenic shock, myocarditis, and isolated right ventricular support for a failing right ventricle in a patient with an LVAD in place.24
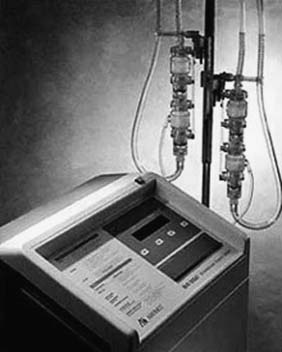
The Abiomed BVS 5000 is one of the most commonly implanted devices for short-term support, typically 5 to 9 days in duration.13,24,27 It is frequently implanted in small, community hospitals to stabilize a patient with cardiogenic shock and to prepare the patient for transfer to a tertiary care center, where the patient can be evaluated for a potential bridge to a longer-term device or cardiac transplantation, or both.13 Favorable elements of this device include its ease of insertion, and postoperative management is uncomplicated enough to eliminate the need for a bedside perfusionist. Many series have published very acceptable results with this device.13,24,27
In 2004, we published a 10-year review of our experience with Abiomed for short-term support in 71 patients.24 This included 19 patients with LVADs, 30 with RVADs, and 22 with BiVADs. Devices were inserted for postcardiotomy shock in 75% of patients and precardiotomy shock in 25% of patients. Mean duration of support was 5 days, and 41% of patients were successfully weaned from mechanical support after myocardial recovery, 11% were bridged to longer-term VADs, and 10% were bridged to transplant. Overall survival on the device was 62%.24
The Abiomed AB5000, the next-generation device after the BVS 5000, is fully automated and has a vacuum-assisted console (Fig. 97-4). Advantages of this device over its predecessor include allowing patients to be more mobile and extending the duration of device support. Preliminary clinical experience with this device has been favorable, although concerns have been raised regarding device-related thrombosis.28
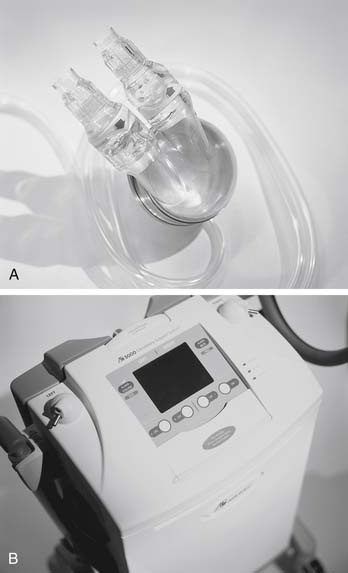
Figure 97–4 Abiomed AB5000. A, AB5000 ventrical pump; B, device console.
(Courtesy of Abiomed, Inc., used with permission.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


