Vascular Wall Biology: Atherosclerosis and Neointimal Hyperplasia
Zhihua Jiang
Scott A. Berceli
Keith C. Ozaki
As a platform for the upcoming chapters that address the management of vascular system disorders, this introductory section summarizes relatively focused aspects of contemporary vascular biology. The emphasis is on the basic science underlying the commonly encountered clinical problem of atherosclerosis, and the typical mechanisms of re-occlusive failure of surgical therapies for atherosclerotic lesions.
Normal Vascular Structure and Function
Early events in the embryology of the vascular system (derived from the mesoderm) lay the foundation for later structure/function relationships. The endothelial cells that line blood vessels are derived from angioblasts, while the smooth muscle cells and fibroblasts that dominate the medial and outer layers are recruited from local mesenchymal cells. During development, strands of these cells cluster and then form cords and tubes. This coalescence of precursor cells into functional blood conduits is called vasculogenesis. These primitive structures then go on to sprout, grow, and remodel to shape the early vascular system. The growth of new endothelial cell-lined tubes from existing blood vessels is called angiogenesis, and this process is observed after birth in multiple clinical scenarios, including wound healing and tumor neovascularization. Finally, hemodynamic forces can drive later outward remodeling of preexisting blood vessels. For instance, arteriogenesis refers to the outward remodeling of pre-existing collateral artery parallel circuits around a hemodynamically significant lesion.
In the typical large- and medium-sized human arteries that are manipulated by vascular surgeons, the wall is organized into three structurally distinct layers. The innermost wall is the intima, and it lies on the luminal surface of the vessel wall in a monolayer of simple squamous endothelial cells. Rather than merely serving as a passive physical barrier separating blood flow from the vascular wall, these cells orchestrate a variety of signals and functions to maintain vascular homeostasis. Endothelial cells actively participate in tissue nutrient and waste exchange, control of intravascular oncotic pressure, coagulation and fibrinolysis, lipid metabolism, and regulation of vascular tone. Through the production and secretion of numerous growth factors and cytokines, they impact surrounding and distant tissues, regulating diverse processes such as inflammatory reactions, vasculogenesis, angiogenesis, and vascular remodeling.
One example of a mediator for endothelial cell regulation is nitric oxide (NO), which is generated in endothelial cells by a constitutively expressed enzyme, endothelial nitric oxide synthase (eNOS), which converts L-arginine to NO and L-citrulline. Using cyclic guanosine 3′, 5′-monophosphate (cGMP) as its second messenger, NO relaxes smooth muscle cells and is thus involved in the regulation of peripheral vascular resistance and hence blood redistribution. In addition to its effect on vasomotor tone, NO inhibits smooth muscle cell proliferation, platelet aggregation, and leukocyte adhesion to the endothelium—early events involved in the pathogenesis of atherosclerosis and restenosis. Heparin, thrombomodulin, prostacyclin (PGI2), and tissue plasminogen activator (TPA) are critical to the normal homeostatic functions of the endothelium. These molecules function together to maintain the nonthrombogenic vascular luminal surface and prevent intravascular coagulation.
Underlying the intimal endothelial cell layer is the internal elastic lamina (IEL), one of several thin sheets of elastin that occupy the tunica media. Arteries differ in the number of elastin layers in the media, and these layers affect the biomechanical properties of the vessel. The media contains layers of circumferentially oriented smooth muscle cells and matrix (collagen and proteoglycans) separated into lamellae by these elastin layers. The outermost elastin layer (external elastic lamina) defines the outer boundary of the media. Smooth muscle cells and extracellular matrix dominate the media’s composition. Muscular arteries can have from 8 to 40 layers of smooth muscle cells in their media. Veins, on the other hand, have a similar wall structure compared to arteries, but a thinner tunica media with few elastin layers. The relaxation or constriction of medial smooth muscle cells in response to stimuli is the primary determinant of the peripheral vascular resistance.
Finally, the adventitia lies immediately adjacent to the external elastic lamina. This layer is composed of loose collagen and elastin fibers, fibroblasts, nerves, and microvessels (vasa vasorum). These microvessels supply nutrients and oxygen to the adventitia and outer media. Fibroblasts are the predominant cell type in the adventitia,
and there are also usually several monocytes.
and there are also usually several monocytes.
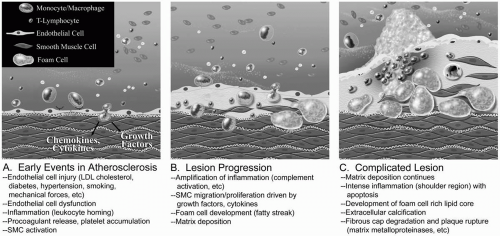 Figure 1-1. Initiation and progression of atherosclerosis. A: Endothelial cell injury disrupts endothelial cell function and leads to changes in permeability, increased leukocyte and platelet adhesive characteristics, and production of cytokines and growth factors that attract leukocytes and drive vascular smooth muscle cell (SMC) migration and proliferation. B: Events such as complement activation further drive the inflammatory response, and activated macrophages phagocytize oxidized low-density lipoprotein (LDL). C: Advanced lesion with increasing amounts of fibrosis, ulceration, calcification, and hemorrhage. (Illustration by John Richardson, Malcom Randall VAMC, Gainesville, FL. Used with permission.) |
Atherosclerosis
Atherosclerosis (athero: gruel or paste; sclerosis: hardness) is a widespread disease process that strikes the intima of large- and medium-sized arteries, leaving deposits of lipids, calcium, and other substances. These plaque lesions become clinically significant when they restrict organ blood flow or rupture to expose their thrombogenic subendothelial tissues, which can lead to acute thrombosis or thromboembolism. Rarely, deep ulcerated lesions can result in arterial wall rupture and hemorrhage. The process is usually segmental (localizes to anatomically distinct locations), allowing local surgical therapies to remove or exclude clinically significant plaques or bypass around such lesions. Atherosclerosis tends to occur in arterial wall areas subjected to disordered flow patterns with low or oscillatory wall shear stress. Epidemiologic risk factors for atherosclerosis include both genetic and environmental forces:
Male gender
Genetic factors
High blood cholesterol (especially lowdensity lipoprotein [LDL] that is higher than 100 mg/dL)
Cigarette smoking and exposure to tobacco smoke
Hypertension
Diabetes mellitus
Obesity
Hyperhomocystinemia
Physical inactivity
These diverse risk factors do not point to an obvious mechanistic pathophysiology. Central pathologic roles have been proposed for lipids, thrombosis, infectious agents (such as cytomegalovirus and Chlamydia pneumoniae), and smooth muscle cells derived from a single progenitor. Recent theories have additionally emphasized inflammatory mechanisms. For instance, C-reactive protein (CRP), which correlates with increased risk for acute cardiac events, is currently under investigation as a risk factor for generalized atherosclerosis. Despite enormous new knowledge over the past half decade, the exact initiators and subsequent molecular mechanisms of atherosclerosis remain undetermined.
Contemporary theories regarding the development of atherosclerosis cite initial endothelial cell injury and endothelial dysfunction (Fig. 1-1). These perturbations in endothelial cell function include:
Changes in permeability
Increased leukocyte and platelet adhesive characteristics (e.g., via upregulation of vascular cell adhesion molecule-1 or VCAM-1)
Production of cytokines and growth factors that attract leukocytes and drive the migration and proliferation of vascular smooth muscle cells and synthesis of new extracellular matrix.
These early lesions may be associated with an accumulation of oxidized LDL in the subendothelial space that is phagocytosed by macrophages that have migrated to the area, leading to foam cells. Some also argue that these initial lesions derive from small accumulations of intimal smooth muscle cells. More mature lesions hold activated macrophages that have phagocytosed lipids, as well as T lymphocytes and smooth muscle cells that have probably migrated into the intima from the underlying media.
Mediators of the clotting cascades have been implicated in the pathophysiology of atherosclerosis. Dysfunctional endothelium shifts from a basal anticoagulant state
to active expression of adhesion molecules, leading to platelet adhesion and aggregation. Platelets, which are known to secrete a number of growth factors and vasoactive substances, adhere to the abnormal endothelium as the lesion progresses. The normal balance of vascular tone dictated by the endothelium may also be pushed toward vasoconstriction with early endothelial cell dysfunction, and these subtle changes can be detected in duplex studies of accessible muscular vessels such as the brachial artery.
to active expression of adhesion molecules, leading to platelet adhesion and aggregation. Platelets, which are known to secrete a number of growth factors and vasoactive substances, adhere to the abnormal endothelium as the lesion progresses. The normal balance of vascular tone dictated by the endothelium may also be pushed toward vasoconstriction with early endothelial cell dysfunction, and these subtle changes can be detected in duplex studies of accessible muscular vessels such as the brachial artery.
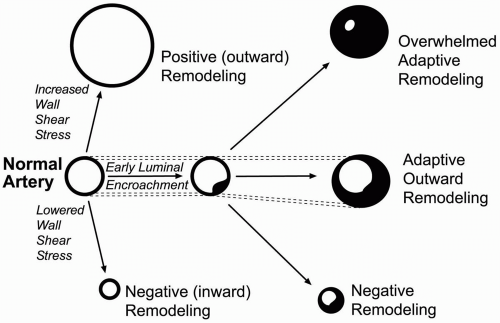 Figure 1-2. Arterial remodeling. In the face of luminal encroachment by an atherosclerotic plaque, adaptive outward remodeling can preserve luminal area. (Illustration by John Richardson, Malcom Randall VAMC, Gainesville, FL. Used with permission.) |
Activation of the complement system additionally drives the inflammatory response, enhancing leukocyte recruitment and activation and smooth muscle cell proliferation. The arterial wall media may not be the only source of smooth muscle cells in the developing atherosclerotic plaque, and some may derive from the adventitia. Circulating progenitor cells also probably participate in the formation of these plaques.
There is a continuous spectrum from the simple fibrofatty lesions to the complicated fibrous plaque, with increasing amounts of fibrosis, ulceration, calcification, and hemorrhage. Development of these lesions is dynamic, with a balance between cell proliferation and apoptosis, and progression usually proceeds over decades. While it is better recognized that the inflammation drives the progression from fatty streak to the advanced atherosclerotic lesion, this chronic inflammation appears to smolder for years before progression to clinically apparent disease. Some evidence shows that with aggressive risk factor modification and statin therapy, or specific lipid-based interventions, lesions may anatomically regress; however, the overwhelming majority of the lesions in this disease are progressive.
As these occlusive lesions develop, the artery wall attempts to accommodate by remodeling (enlarging overall dimensions to maintain lumen caliber). Arteries maintain a general ability to reshape in response to hemodynamic and biochemical stimuli (Fig. 1-2




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
