OBJECTIVES
The student understands the general mechanisms involved in local vascular control:
![]() Identifies the major ways in which smooth muscle differs anatomically and functionally from striated muscle.
Identifies the major ways in which smooth muscle differs anatomically and functionally from striated muscle.
![]() Lists the steps leading to cross-bridge cycling in smooth muscle.
Lists the steps leading to cross-bridge cycling in smooth muscle.
![]() Lists the major ion channels involved in the regulation of membrane potential in smooth muscle.
Lists the major ion channels involved in the regulation of membrane potential in smooth muscle.
![]() Describes the processes of electromechanical and pharmacomechanical coupling in smooth muscle.
Describes the processes of electromechanical and pharmacomechanical coupling in smooth muscle.
![]() Defines basal tone.
Defines basal tone.
![]() Lists several substances potentially involved in local metabolic control.
Lists several substances potentially involved in local metabolic control.
![]() States the local metabolic vasodilator hypothesis.
States the local metabolic vasodilator hypothesis.
![]() Describes how vascular tone may be influenced by endothelin, prostaglandins, histamine, and bradykinin.
Describes how vascular tone may be influenced by endothelin, prostaglandins, histamine, and bradykinin.
![]() Describes the myogenic response of blood vessels.
Describes the myogenic response of blood vessels.
![]() Defines active and reactive hyperemia and indicates a possible mechanism for each.
Defines active and reactive hyperemia and indicates a possible mechanism for each.
![]() Defines autoregulation of blood flow and briefly describes the metabolic, myogenic, and tissue pressure theories of autoregulation.
Defines autoregulation of blood flow and briefly describes the metabolic, myogenic, and tissue pressure theories of autoregulation.
![]() Defines neurogenic tone of vascular muscle and describes how sympathetic neural influences can alter it.
Defines neurogenic tone of vascular muscle and describes how sympathetic neural influences can alter it.
![]() Describes how vascular tone is influenced by circulating catecholamines, vasopressin, and angiotensin II.
Describes how vascular tone is influenced by circulating catecholamines, vasopressin, and angiotensin II.
![]() Lists the major influences on venous diameters.
Lists the major influences on venous diameters.
![]() Describes how control of flow differs between organs with strong local metabolic control of arteriolar tone and organs with strong neurogenic control of arteriolar tone.
Describes how control of flow differs between organs with strong local metabolic control of arteriolar tone and organs with strong neurogenic control of arteriolar tone.
The student knows the dominant mechanisms of flow and blood volume control in the major body organs:
![]() States the relative importance of local metabolic and neural control of coronary blood flow.
States the relative importance of local metabolic and neural control of coronary blood flow.
![]() Defines systolic compression and indicates its relative importance to blood flow in the endocardial and epicardial regions of the right and left ventricular walls.
Defines systolic compression and indicates its relative importance to blood flow in the endocardial and epicardial regions of the right and left ventricular walls.
![]() Describes the major mechanisms of flow and blood volume control in each of the following systemic organs: skeletal muscle, brain, splanchnic organs, kidney, and skin.
Describes the major mechanisms of flow and blood volume control in each of the following systemic organs: skeletal muscle, brain, splanchnic organs, kidney, and skin.
![]() States why mean pulmonary arterial pressure is lower than mean systemic arterial pressure.
States why mean pulmonary arterial pressure is lower than mean systemic arterial pressure.
![]() Describes how pulmonary vascular control differs from that in systemic organs.
Describes how pulmonary vascular control differs from that in systemic organs.
![]() Because the body’s metabolic needs are continually changing, the cardiovascular system must continually make adjustments in the diameter of its vessels. The purposes of these vascular changes are (1) to efficiently distribute the cardiac output among tissues with different current needs (the job of arterioles) and (2) to regulate the distribution of blood volume and cardiac filling (the job of veins). In this chapter, we discuss our current understanding of how all this is accomplished.
Because the body’s metabolic needs are continually changing, the cardiovascular system must continually make adjustments in the diameter of its vessels. The purposes of these vascular changes are (1) to efficiently distribute the cardiac output among tissues with different current needs (the job of arterioles) and (2) to regulate the distribution of blood volume and cardiac filling (the job of veins). In this chapter, we discuss our current understanding of how all this is accomplished.
VASCULAR SMOOTH MUSCLE
![]() Although long-term adaptations in vascular diameters may depend on remodeling of both the active (ie, smooth muscle) and passive (ie, structural, connective tissue) components of the vascular wall, short-term vascular diameter adjustments are made by regulating the contractile activity of vascular smooth muscle cells. These contractile cells are present in the walls of all vessels except capillaries. The task of the vascular smooth muscle is unique, because to maintain a certain vessel diameter in the face of the continual distending pressure of the blood within it, the vascular smooth muscle must be able to sustain active tension for prolonged periods.
Although long-term adaptations in vascular diameters may depend on remodeling of both the active (ie, smooth muscle) and passive (ie, structural, connective tissue) components of the vascular wall, short-term vascular diameter adjustments are made by regulating the contractile activity of vascular smooth muscle cells. These contractile cells are present in the walls of all vessels except capillaries. The task of the vascular smooth muscle is unique, because to maintain a certain vessel diameter in the face of the continual distending pressure of the blood within it, the vascular smooth muscle must be able to sustain active tension for prolonged periods.
There are many functional characteristics that distinguish smooth muscle from either skeletal or cardiac muscle. For example, when compared with these other muscle types, smooth muscle cells
1. contract and relax much more slowly;
2. can change their contractile activity as a result of either action potentials or changes in resting membrane potential;
3. can change their contractile activity in the absence of any changes in membrane potential;
4. can maintain tension for prolonged periods at low energy cost; and
5. can be activated by stretch.
Vascular smooth muscle cells are small (approximately 5 μm × 50 μm) spindle-shaped cells, usually arranged circumferentially or at small helical angles in muscular blood vessel walls. In many, but not all, vessels, adjacent smooth muscle cells are electrically connected by gap junctions similar to those found in the myocardium.
Contractile Processes
![]() Just as in other muscle types, smooth muscle force development and shortening are thought to be the result of cross-bridge interaction between thick and thin contractile filaments composed of myosin and actin, respectively. In smooth muscle, however, these filaments are not arranged in regular, repeating sarcomere units. As a consequence, “smooth” muscle cells lack the microscopically visible striations, characteristic of skeletal and cardiac muscle cells. The actin filaments in smooth muscle are much longer than those in striated muscle. Many of these actin filaments attach to the inner surface of the cell at structures called dense bands. In the interior of the cell, actin filaments do not attach to Z lines but rather anchor to small transverse structures called dense bodies that are themselves tethered to the surface membrane by cable-like intermediate filaments. Myosin filaments are interspersed between the smooth muscle actin filaments but in a more haphazard fashion than the regular interweaving pattern of striated muscle. In striated muscle, the contractile filaments are invariably aligned with the long axis of the cell, whereas in smooth muscle, many contractile filaments travel obliquely or even transversely to the long axis of the cell. Despite the absence of organized sarcomeres, changes in smooth muscle length affect its ability to actively develop tension. That is, smooth muscle exhibits a “length–tension relationship” analogous to that observed in striated muscle (see, Figure 2–8). As in striated muscle, the strength of the cross-bridge interaction between myosin and actin filaments in smooth muscle is controlled primarily by changes in the intracellular free Ca2+ level, which range from approximately 10-8 M in the relaxed muscle to 10-5 M during maximal contraction. However, the sequence of steps linking an increased free Ca2+ concentration to contractile filament interaction is different in smooth muscle than in striated muscle. In the smooth muscle:
Just as in other muscle types, smooth muscle force development and shortening are thought to be the result of cross-bridge interaction between thick and thin contractile filaments composed of myosin and actin, respectively. In smooth muscle, however, these filaments are not arranged in regular, repeating sarcomere units. As a consequence, “smooth” muscle cells lack the microscopically visible striations, characteristic of skeletal and cardiac muscle cells. The actin filaments in smooth muscle are much longer than those in striated muscle. Many of these actin filaments attach to the inner surface of the cell at structures called dense bands. In the interior of the cell, actin filaments do not attach to Z lines but rather anchor to small transverse structures called dense bodies that are themselves tethered to the surface membrane by cable-like intermediate filaments. Myosin filaments are interspersed between the smooth muscle actin filaments but in a more haphazard fashion than the regular interweaving pattern of striated muscle. In striated muscle, the contractile filaments are invariably aligned with the long axis of the cell, whereas in smooth muscle, many contractile filaments travel obliquely or even transversely to the long axis of the cell. Despite the absence of organized sarcomeres, changes in smooth muscle length affect its ability to actively develop tension. That is, smooth muscle exhibits a “length–tension relationship” analogous to that observed in striated muscle (see, Figure 2–8). As in striated muscle, the strength of the cross-bridge interaction between myosin and actin filaments in smooth muscle is controlled primarily by changes in the intracellular free Ca2+ level, which range from approximately 10-8 M in the relaxed muscle to 10-5 M during maximal contraction. However, the sequence of steps linking an increased free Ca2+ concentration to contractile filament interaction is different in smooth muscle than in striated muscle. In the smooth muscle:
1. Intracellular free Ca2+ first forms a complex with the calcium-binding protein calmodulin.
2. The Ca2+–calmodulin complex then activates a phosphorylating enzyme called myosin light-chain kinase (MLC kinase).
3. This enzyme allows the phosphorylation by adenosine triphosphate (ATP) of the light-chain protein that is a portion of the cross-bridge head of myosin (MLC).
4. MLC phosphorylation enables cross-bridge formation and cycling during which energy from ATP is utilized for tension development and shortening.
Smooth muscle is also unique in that once tension is developed, it can be maintained at very low energy costs, that is, without the need to continually split ATP in cross-bridge cycling. The mechanisms responsible are still somewhat unclear but presumably involve very slowly cycling or even noncycling cross-bridges. This is often referred to as the latch state and may involve light-chain dephosphorylation of attached cross-bridges.
By mechanisms that are yet incompletely understood, it is apparent that vascular smooth muscle contractile activity is regulated not only by changes in intracellular free Ca2+ levels but also by changes in the Ca2+ sensitivity of the contractile machinery. Thus, the contractile state of vascular smooth muscle may sometimes change in the absence of changes in intracellular free Ca2+ levels. In part, this apparently variable Ca2+ sensitivity of the activation of smooth muscle contractile apparatus may be due to the variable activity of another enzyme, myosin phosphatase, that facilitates some reaction that involves the phosphorylated MLC as a reactant. For example, factors that increase the intracellular concentrations of cyclic nucleotides often lead to relaxation of the vascular smooth muscle. Thus, the net state of phosphorylation of the MLC (and thus presumably contractile strength) depends on some sort of balance between the effects of the Ca2+-dependent enzyme MLC kinase, and the Ca2+-independent enzyme MLC phosphatase.1
Membrane Potentials
Smooth muscle cells have resting membrane potentials ranging from –40 to –65 mV and thus are generally less negative than those in striated muscle. As in all cells, the resting membrane potential of the smooth muscle is determined largely by the cell permeability to potassium. Many types of K+ channels have been identified in smooth muscle. The one that seems to be predominantly responsible for determining the resting membrane potential is termed an inward rectifying-type K+ channel. There are also ATP-dependent K+ channels that are closed when cellular ATP levels are normal but open if ATP levels fall. Such channels have been proposed to be important in matching organ blood flow to the metabolic state of the tissue.
Smooth muscle cells regularly have action potentials only in certain vessels. When they do occur, smooth muscle action potentials are initiated primarily by inward Ca2+ current and are developed slowly like the “slow-type” cardiac action potentials (see Figures 2–2C and D). As in the heart, this inward (depolarizing) Ca2+ current flows through a voltage-operated channel (VOC) for Ca2+; this type of channel is one of several types of calcium channels present in the smooth muscle. The repolarization phase of the action potential occurs primarily by an outward flux of potassium ions through both delayed K+ channels and calcium-activated K+ channels.
Many types of ion channels in addition to those mentioned have been identified in vascular smooth muscle, but in most cases, their exact role in cardiovascular function remains obscure. For example, there appear to be nonselective, stretch-sensitive cation channels that may be involved in the response of smooth muscle to stretch. The reader should note, however, that many of the important ion channels in vascular smooth muscle are also important in heart muscle (see Table 2–1).
Electromechanical versus Pharmacomechanical Coupling
![]() In smooth muscle, changes in intracellular free Ca2+ levels can occur both with and without changes in membrane potential. The processes involved are called electromechanical coupling and pharmacomechanical coupling, respectively, and are illustrated in Figure 7–1.
In smooth muscle, changes in intracellular free Ca2+ levels can occur both with and without changes in membrane potential. The processes involved are called electromechanical coupling and pharmacomechanical coupling, respectively, and are illustrated in Figure 7–1.
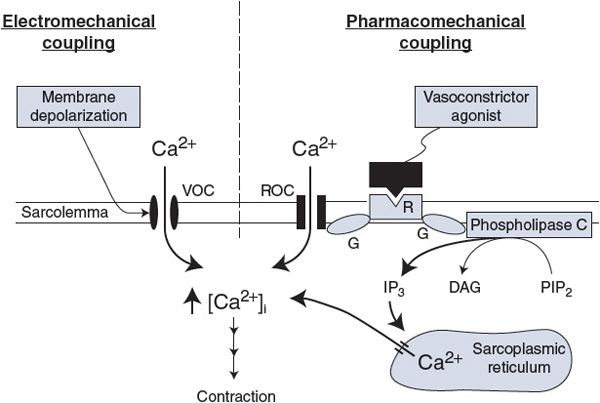
Figure 7–1. General mechanisms for activation of the vascular smooth muscle. VOC, voltage-operated Ca2+ channel; ROC, receptor-operated Ca2+ channel; R, agonist-specific receptor; G, GTP-binding protein; PIP2, phosphatidylinositol biphosphate; IP3, inositol triphosphate; DAG, diacylglycerol.
Electromechanical coupling, shown in the left half of Figure 7–1, occurs because the smooth muscle surface membrane contains VOCs for calcium (the same VOCs that are involved in action potential generation). Membrane depolarization increases the open-state probability of these channels and thus leads to smooth muscle cell contraction and vessel constriction. Conversely, membrane hyperpolarization leads to smooth muscle relaxation and vessel dilation. Because the VOCs for Ca2+ are partially activated by the low resting membrane potential of the vascular smooth muscle, changes in resting potential can alter the resting calcium influx rate and therefore the basal contractile state.
With pharmacomechanical coupling, chemical agents (eg, released neurotransmitters) can induce smooth muscle contraction without the need for a change in membrane potential. As illustrated on the right side of Figure 7–1, the combination of a vasoconstrictor agonist (such as norepinephrine) with a specific membrane-bound receptor (such as an α1-adrenergic receptor) initiates events that cause intracellular free Ca2+ levels to increase for two reasons. One, the activated receptor may open surface membrane receptor-operated channels for Ca2+ that allow Ca2+ influx from the extracellular fluid. Two, the activated receptor may induce the formation of an intracellular “second messenger,” inositol trisphosphate (IP3), which opens specific channels that release Ca2+ from the intracellular sarcoplasmic reticulum stores. In both processes, the activated receptor first stimulates specific guanosine triphosphate-binding proteins (GTP-binding proteins or G proteins). Such receptor-associated G proteins seem to represent a general first step through which most membrane receptors operate to initiate their particular cascade of events that ultimately lead to specific cellular responses.
The reader should not conclude from Figure 7–1 that all vasoactive chemical agents (chemical agents that cause vascular effects) produce their actions on the smooth muscle without changing membrane potential. In fact, most vasoactive chemical agents do cause changes in membrane potential because their receptors can be linked, by G proteins or other means, to ion channels of many kinds.
Not shown in Figure 7–1 are the processes that remove Ca2+ from the cytoplasm of the vascular smooth muscle, although they are important as well in determining the free cytosolic Ca2+ levels. As in cardiac cells (see Figure 2–7), smooth muscle cells actively pump calcium into the sarcoplasmic reticulum and outward across the sarcolemma. Calcium is also countertransported out of the cell in exchange for sodium.
Mechanisms for Relaxation
Hyperpolarization of the cell membrane is one mechanism for causing smooth muscle relaxation and vessel dilation. In addition, however, there are at least two general mechanisms by which certain chemical vasodilator agents can cause smooth muscle relaxation by pharmacomechanical means. In Figure 7–1, the specific receptor for a chemical vasoconstrictor agent is shown linked by a specific G protein to phospholipase C. In an analogous manner, other specific receptors may be linked by other specific G proteins to other enzymes that produce second messengers other than IP3. An example is the β2-adrenergic receptor2 that is present in arterioles of the skeletal muscle and liver. β2-Receptors are not innervated but can sometimes be activated by abnormally elevated levels of circulating epinephrine. The β2-receptor is linked by a particular G protein (Gs) to adenylate cyclase. Adenylate cyclase catalyzes the conversion of ATP to cyclic adenosine monophosphate (cAMP). Increased intracellular levels of cAMP cause the activation of protein kinase A, a phosphorylating enzyme that in turn causes phosphorylation of proteins at many sites. The overall result is stimulation of Ca2+ efflux, membrane hyperpolarization, and decreased contractile machinery sensitivity to Ca2+—all of which act synergistically to cause vasodilation. In addition to epinephrine, histamine and vasoactive intestinal peptide are other vasodilator substances that act through the cAMP pathway.
In addition to cAMP, cyclic guanosine monophosphate (cGMP) is an important intracellular second messenger that causes vascular smooth muscle relaxation. Nitric oxide is an important vasodilator substance that operates via the cGMP pathway. Nitric oxide can be produced by endothelial cells and also by nitrates, a clinically important class of vasodilator drugs. Nitric oxide is gaseous and easily diffuses into smooth muscle cells, where it activates the enzyme guanylyl cyclase that in turn causes cGMP formation.
CONTROL OF ARTERIOLAR TONE
![]() Vascular tone is a term commonly used to characterize the general contractile state of a vessel or a vascular region. The “vascular tone” of a region can be taken as an indication of the “level of activation” of the individual smooth muscle cells in that region. As described in Chapter 6, the blood flow through any organ is determined largely by its vascular resistance, which is dependent primarily on the diameter of its arterioles. Consequently, an organ’s flow is controlled by factors that influence the arteriolar smooth muscle tone.
Vascular tone is a term commonly used to characterize the general contractile state of a vessel or a vascular region. The “vascular tone” of a region can be taken as an indication of the “level of activation” of the individual smooth muscle cells in that region. As described in Chapter 6, the blood flow through any organ is determined largely by its vascular resistance, which is dependent primarily on the diameter of its arterioles. Consequently, an organ’s flow is controlled by factors that influence the arteriolar smooth muscle tone.
Basal Tone
Arterioles remain in a state of partial constriction even when all external influences on them are removed; hence, they are said to have a degree of basal tone (sometimes referred to as intrinsic tone). The understanding of the mechanism is incomplete, but basal arteriolar tone may be a reflection of the fact that smooth muscle cells inherently and actively resist being stretched as they continually are in pressurized arterioles. Another hypothesis is that the basal tone of arterioles is the result of a tonic production of local vasoconstrictor substances by the endothelial cells that line their inner surface. In any case, this basal tone establishes a baseline of partial arteriolar constriction from which the external influences on arterioles exert their dilating or constricting effects. These influences can be separated into three categories: local influences, neural influences, and hormonal influences.
Local Influences on Arterioles
METABOLIC INFLUENCES
![]() The arterioles that control flow through a given organ lie within the organ tissue itself. Thus, arterioles and the smooth muscle in their walls are exposed to the chemical composition of the interstitial fluid of the organ they serve. The interstitial concentrations of many substances reflect the balance between the metabolic activity of the tissue and its blood supply. Interstitial oxygen levels, for example, fall whenever the tissue cells are using oxygen faster than it is being supplied to the tissue by blood flow. Conversely, interstitial oxygen levels rise whenever excess oxygen is being delivered to a tissue from the blood. In nearly all vascular beds, exposure to low oxygen reduces arteriolar tone and causes vasodilation, whereas high oxygen levels cause arteriolar vasoconstriction.3 Thus, a local feedback mechanism exists that automatically operates on arterioles to regulate a tissue’s blood flow in accordance with its metabolic needs. Whenever blood flow and oxygen delivery fall below a tissue’s oxygen demand, the oxygen levels around arterioles fall, the arterioles dilate, and the blood flow through the organ appropriately increases.
The arterioles that control flow through a given organ lie within the organ tissue itself. Thus, arterioles and the smooth muscle in their walls are exposed to the chemical composition of the interstitial fluid of the organ they serve. The interstitial concentrations of many substances reflect the balance between the metabolic activity of the tissue and its blood supply. Interstitial oxygen levels, for example, fall whenever the tissue cells are using oxygen faster than it is being supplied to the tissue by blood flow. Conversely, interstitial oxygen levels rise whenever excess oxygen is being delivered to a tissue from the blood. In nearly all vascular beds, exposure to low oxygen reduces arteriolar tone and causes vasodilation, whereas high oxygen levels cause arteriolar vasoconstriction.3 Thus, a local feedback mechanism exists that automatically operates on arterioles to regulate a tissue’s blood flow in accordance with its metabolic needs. Whenever blood flow and oxygen delivery fall below a tissue’s oxygen demand, the oxygen levels around arterioles fall, the arterioles dilate, and the blood flow through the organ appropriately increases.
Many substances in addition to oxygen are present within tissues and can affect the tone of the vascular smooth muscle. When the metabolic rate of skeletal muscle is increased by exercise, tissue levels of oxygen decrease, but those of carbon dioxide, H+, and K+ increase. Muscle tissue osmolarity also increases during exercise. All these chemical alterations cause arteriolar dilation. In addition, with increased metabolic activity or oxygen deprivation, cells in many tissues may release adenosine, which is an extremely potent vasodilator agent.
At present, it is not known which of these (and possibly other) metabolically related chemical alterations within tissues are most important in the local metabolic control of blood flow. It appears likely that arteriolar tone depends on the combined action of many factors.
For conceptual purposes, Figure 7–2 summarizes current understanding of local metabolic control. Vasodilator factors enter the interstitial space from the tissue cells at a rate proportional to tissue metabolism. These vasodilator factors are removed from the tissue at a rate proportional to blood flow. Whenever tissue metabolism is proceeding at a rate for which the blood flow is inadequate, the interstitial vasodilator factor concentrations automatically build up and cause the arterioles to dilate. This, of course, causes blood flow to increase. The process continues until blood flow has risen sufficiently to appropriately match the tissue metabolic rate and prevent further accumulation of vasodilator factors. The same system also operates to reduce blood flow when it is higher than required by the tissue’s metabolic activity, because this situation causes a reduction in the interstitial concentrations of metabolic vasodilator factors.
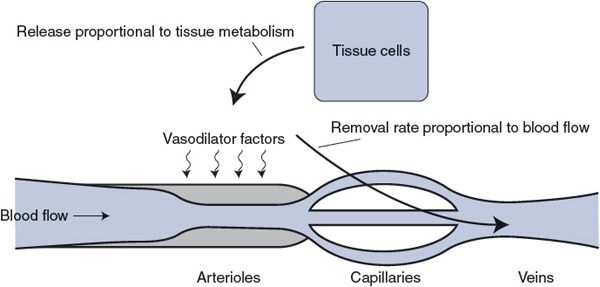
Figure 7–2. Local metabolic vasodilator hypothesis.
Local metabolic mechanisms represent by far the most important means of local flow control. By these mechanisms, individual organs are able to regulate their own flow in accordance with their specific metabolic needs.
As indicated below, several other types of local influences on blood vessels have been identified. However, many of these represent fine-tuning mechanisms and many are important only in certain, usually pathological, situations.
LOCAL INFLUENCES FROM ENDOTHELIAL CELLS
Endothelial cells cover the entire inner surface of the cardiovascular system. A large number of studies have shown that blood vessels respond very differently to certain vascular influences when their endothelial lining is missing. Acetylcholine, for example, causes vasodilation of intact vessels but causes vasoconstriction of vessels stripped of their endothelial lining. This and similar results led to the realization that endothelial cells can actively participate in the control of arteriolar diameter by producing local chemicals that affect the tone of the surrounding smooth muscle cells. In the case of the vasodilator effect of infusing acetylcholine through intact vessels, the vasodilator influence produced by endothelial cells has been identified as nitric oxide. Nitric oxide is produced within endothelial cells from the amino acid, L-arginine, by the action of an enzyme, nitric oxide synthase. Nitric oxide synthase is activated by a rise in the intracellular level of the Ca2+. Nitric oxide is a small lipid-soluble molecule that, once formed, easily diffuses into adjacent smooth muscle cells where it causes relaxation by stimulating cGMP production as mentioned previously.
Acetylcholine and several other agents (including bradykinin, vasoactive intestinal peptide, and substance P) stimulate endothelial cell nitric oxide production because their receptors on endothelial cells are linked to receptor-operated Ca2+ channels. Probably more importantly from a physiological standpoint, flow-related shear stresses on endothelial cells stimulate their nitric oxide production presumably because stretch-sensitive channels for Ca2+ are activated. Such flow-related endothelial cell nitric oxide production may explain why, for example, exercise and increased blood flow through muscles of the lower leg can cause dilation of the blood-supplying femoral artery at points far upstream of the exercising muscle itself.
Agents that block nitric oxide production by inhibiting nitric oxide synthase cause significant increases in the vascular resistances of most organs. For this reason, it is believed that endothelial cells are normally always producing some nitric oxide that is importantly involved, along with other factors, in reducing the normal resting tone of arterioles throughout the body.
Endothelial cells have also been shown to produce several other locally acting vasoactive agents including the vasodilators “endothelial-derived hyperpolarizing factor”, prostacyclin and the vasoconstrictor endothelin. Endothelin in particular is the topic of intense current research. It has the greatest vasoconstrictor potency of any known substance and appears to have many other biological effects as well. Much recent evidence suggests that endothelin may play important roles in such important overall process such as bodily salt handling and blood pressure regulation.
One general unresolved issue with the concept that arteriolar tone (and therefore local nutrient blood flow) is regulated by factors produced by arteriolar endothelial cells is how these cells could know what the metabolic needs of the downstream tissue are. This is because the endothelial cells lining arterioles are exposed to arterial blood whose composition is constant regardless of flow rate or what is happening downstream. One hypothesis is that there exists some sort of communication system between vascular endothelial cells. That way, endothelial cells in capillaries or venules could telegraph upstream information about whether the blood flow is indeed adequate.
OTHER LOCAL CHEMICAL INFLUENCES
![]() In addition to local metabolic influences on vascular tone, many specific locally-produced and locally-reacting chemical substances have been identified that have vascular effects and therefore could be important in local vascular regulation in certain instances. In most cases, however, definite information about the relative importance of these substances in cardiovascular regulation is lacking.
In addition to local metabolic influences on vascular tone, many specific locally-produced and locally-reacting chemical substances have been identified that have vascular effects and therefore could be important in local vascular regulation in certain instances. In most cases, however, definite information about the relative importance of these substances in cardiovascular regulation is lacking.
Prostaglandins and thromboxane are a group of several chemically related products of the cyclooxygenase pathway of arachidonic acid metabolism. Certain prostaglandins are potent vasodilators, whereas others are potent vasoconstrictors. Despite the vasoactive potency of the prostaglandins and the fact that most tissues (including endothelial cells and vascular smooth muscle cells) are capable of synthesizing prostaglandins, it has not been demonstrated convincingly that prostaglandins play a crucial role in normal vascular control. It is clear, however, that vasodilator prostaglandins are involved in inflammatory responses. Consequently, inhibitors of prostaglandin synthesis, such as aspirin, are effective anti-inflammatory drugs. Prostaglandins produced by platelets and endothelial cells are important in the hemostatic (flow stopping, antibleeding) vasoconstrictor and platelet-aggregating responses to vascular injury. Hence, aspirin is often prescribed to reduce the tendency for blood clotting–especially in patients with potential coronary flow limitations. Arachidonic acid metabolites produced via the lipoxygenase system (eg, leukotrienes) also have vasoactive properties and may influence blood flow and vascular permeability during inflammatory processes.
Histamine is synthesized and stored in high concentrations in secretory granules of tissue mast cells and circulating basophils. When released, histamine produces arteriolar vasodilation (via the cAMP pathway) and increases vascular permeability, which leads to edema formation and local tissue swelling. Histamine increases vascular permeability by causing separations in the junctions between the endothelial cells that line the vascular system. Histamine release is classically associated with antigen–antibody reactions in various allergic and immune responses. Many drugs and physical or chemical insults that damage tissue also cause histamine release. Histamine can stimulate sensory nerve endings to cause itching and pain sensations. Although clearly important in many pathological situations, it seems unlikely that histamine participates in normal cardiovascular regulation.
Bradykinin is a small polypeptide that has approximately ten times the vasodilator potency of histamine on a molar basis. It also acts to increase capillary permeability by opening the junctions between endothelial cells. Bradykinin is formed from certain plasma globulin substrates by the action of an enzyme, kallikrein, and is subsequently rapidly degraded into inactive fragments by various tissue kinases. Like histamine, bradykinin is thought to be involved in the vascular responses associated with tissue injury and immune reactions. It also stimulates nociceptive nerves and may thus be involved in the pain associated with tissue injury.
TRANSMURAL PRESSURE
The passive elastic mechanical properties of arteries and veins and how changes in transmural pressure affect their diameters are discussed in Chapter 6. The effect of transmural pressure on arteriolar diameter is more complex because arterioles respond both passively and actively to changes in transmural pressure. For example, a sudden increase in the internal pressure within an arteriole produces (1) first an initial slight passive mechanical distention (slight because arterioles are relatively thick-walled and muscular), and (2) then an active constriction that, within seconds, may completely reverse the initial distention. A sudden decrease in transmural pressure elicits essentially the opposite response, that is, an immediate passive decrease in diameter followed shortly by a decrease in active tone, which returns the arteriolar diameter to near that which existed before the pressure change. The active phase of such behavior is referred to as a myogenic response, because it seems to originate within the smooth muscle itself. The mechanism of the myogenic response is not known for certain, but stretch-sensitive ion channels on arteriolar vascular smooth muscle cells are likely candidates for involvement.
All arterioles have some normal distending pressure to which they are probably actively responding. Therefore, the myogenic mechanism is likely to be a fundamentally important factor in determining the basal tone of arterioles everywhere. Also, for obvious reasons and as soon discussed, the myogenic response is potentially involved in the vascular reaction to any cardiovascular disturbance that involves a change in arteriolar transmural pressure.
FLOW RESPONSES CAUSED BY LOCAL MECHANISMS
Active Hyperemia–In organs with a highly variable metabolic rate, such as skeletal and cardiac muscles, the blood flow closely follows the tissue’s metabolic rate. For example, skeletal muscle blood flow increases within seconds of the onset of muscle exercise and returns to control values shortly after exercise ceases. This phenomenon, which is illustrated in Figure 7–3A, is known as exercise or active hyperemia (hyperemia means high flow). It should be clear how active hyperemia could result from the local metabolic vasodilator feedback on the arteriolar smooth muscle. As alluded to previously, once initiated by local metabolic influences on small resistance vessels, endothelial flow-dependent mechanisms may assist in propagating the vasodilation to larger vessels upstream, which helps promote the delivery of blood to the exercising muscle.
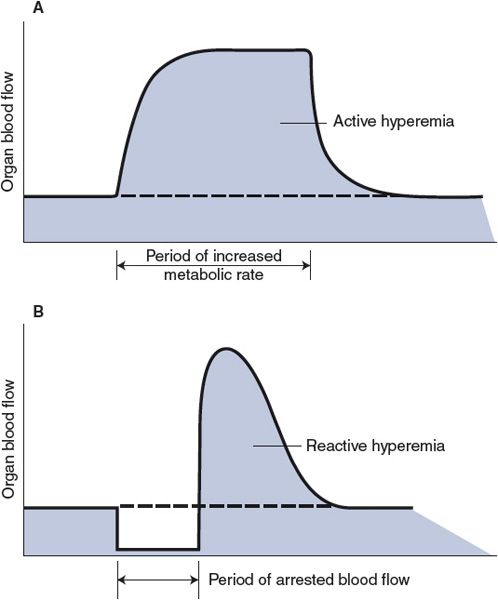
Figure 7–3. Organ blood flow responses caused by local mechanisms: active and reactive hyperemias.
Reactive Hyperemia—In this case, the higher-than-normal blood flow occurs transiently after the removal of any restriction that has caused a period of lower-than-normal blood flow and is sometimes referred to as postocclusion hyperemia. The phenomenon is illustrated in Figure 7–3B. For example, flow through an extremity is higher than normal for a period after a tourniquet is removed from the extremity. Both local metabolic and myogenic mechanisms may be involved in producing reactive hyperemia. The magnitude and duration of reactive hyperemia depend on the duration and severity of the occlusion as well as the metabolic rate of the tissue. These findings are best explained by an interstitial accumulation of metabolic vasodilator substances during the period of flow restriction. However, unexpectedly large flow increases can follow arterial occlusions lasting only 1 or 2 s. These may be explained best by a myogenic dilation response to the reduced intravascular pressure and decreased stretch of the arteriolar walls that exists during the period of occlusion.
Autoregulation—Except when displaying active and reactive hyperemia, nearly all organs tend to keep their blood flow constant despite variations in arterial pressure—that is, they autoregulate their blood flow. As shown in Figure 7–4A, an abrupt increase in arterial pressure is normally accompanied by an initial abrupt increase in organ blood flow that then gradually returns toward normal despite the sustained elevation in arterial pressure. The initial rise in flow with increased pressure is expected from the basic flow equation (![]() = ΔP/R). The subsequent return of flow toward the normal level is caused by a gradual increase in active arteriolar tone and resistance to blood flow. Ultimately, a new steady state is reached with only slightly elevated blood flow because the increased driving pressure is counteracted by a higher-than-normal vascular resistance. As with the phenomenon of reactive hyperemia, blood flow autoregulation may be caused by both local metabolic feedback mechanisms and myogenic mechanisms. The arteriolar vasoconstriction responsible for the autoregulatory response shown in Figure 7–4A, for example, may be partially due to (1) a “washout” of metabolic vasodilator factors from the interstitium by the excessive initial blood flow and (2) a myogenic increase in arteriolar tone stimulated by the increase in stretching forces that the increase in pressure imposes on the vessel walls. There is also a tissue pressure hypothesis of blood flow autoregulation for which it is assumed that an abrupt increase in arterial pressure causes transcapillary fluid filtration and thus leads to a gradual increase in interstitial fluid volume and pressure. Presumably the increase in extravascular pressure would cause a decrease in vessel diameter by simple compression. This mechanism might be especially important in organs such as the kidney and brain whose volumes are constrained by external structures.
= ΔP/R). The subsequent return of flow toward the normal level is caused by a gradual increase in active arteriolar tone and resistance to blood flow. Ultimately, a new steady state is reached with only slightly elevated blood flow because the increased driving pressure is counteracted by a higher-than-normal vascular resistance. As with the phenomenon of reactive hyperemia, blood flow autoregulation may be caused by both local metabolic feedback mechanisms and myogenic mechanisms. The arteriolar vasoconstriction responsible for the autoregulatory response shown in Figure 7–4A, for example, may be partially due to (1) a “washout” of metabolic vasodilator factors from the interstitium by the excessive initial blood flow and (2) a myogenic increase in arteriolar tone stimulated by the increase in stretching forces that the increase in pressure imposes on the vessel walls. There is also a tissue pressure hypothesis of blood flow autoregulation for which it is assumed that an abrupt increase in arterial pressure causes transcapillary fluid filtration and thus leads to a gradual increase in interstitial fluid volume and pressure. Presumably the increase in extravascular pressure would cause a decrease in vessel diameter by simple compression. This mechanism might be especially important in organs such as the kidney and brain whose volumes are constrained by external structures.
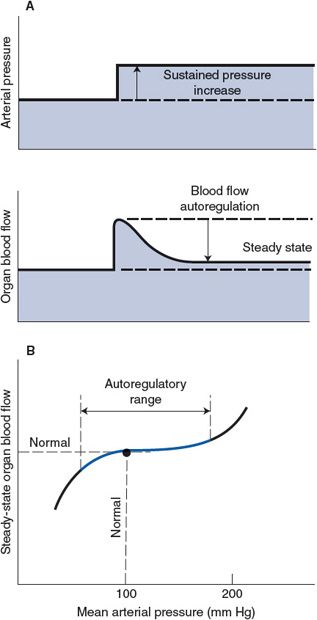
Figure 7–4. Autoregulation of organ blood flow.
Although not illustrated in Figure 7–4A, autoregulatory mechanisms operate in the opposite direction in response to a decrease in arterial pressure below the normal value. One important general consequence of local autoregulatory mechanisms is that the steady-state blood flow in many organs tends to remain near the normal value over quite a wide range of arterial pressure. This is illustrated in the graph in Figure 7–4B. As discussed later, the inherent ability of certain organs to maintain adequate blood flow despite lower-than-normal arterial pressure is of considerable importance in situations such as shock from blood loss.
Neural Influences on Arterioles
SYMPATHETIC VASOCONSTRICTOR NERVES
![]() These neural fibers innervate arterioles in all systemic organs and provide by far the most important means of reflex control of the vasculature. Sympathetic vasoconstrictor nerves are the backbone of the system for controlling total peripheral resistance and are thus essential participants in global cardiovascular tasks such as regulating arterial blood pressure.
These neural fibers innervate arterioles in all systemic organs and provide by far the most important means of reflex control of the vasculature. Sympathetic vasoconstrictor nerves are the backbone of the system for controlling total peripheral resistance and are thus essential participants in global cardiovascular tasks such as regulating arterial blood pressure.
![]() Sympathetic vasoconstrictor nerves release norepinephrine from their terminal structures in amounts generally proportional to their action potential frequency. Norepinephrine causes an increase in the tone of arterioles after combining with an α1-adrenergic receptor on smooth muscle cells. Norepinephrine appears to increase vascular tone primarily by pharmacomechanical means. The mechanism involves G-protein linkage of α-adrenergic receptors to phospholipase C and subsequent Ca2+ release from intracellular stores by the action of the second messenger IP3, as illustrated on the right side of Figure 7–1.
Sympathetic vasoconstrictor nerves release norepinephrine from their terminal structures in amounts generally proportional to their action potential frequency. Norepinephrine causes an increase in the tone of arterioles after combining with an α1-adrenergic receptor on smooth muscle cells. Norepinephrine appears to increase vascular tone primarily by pharmacomechanical means. The mechanism involves G-protein linkage of α-adrenergic receptors to phospholipase C and subsequent Ca2+ release from intracellular stores by the action of the second messenger IP3, as illustrated on the right side of Figure 7–1.
Sympathetic vasoconstrictor nerves normally have a continual or tonic firing activity. This tonic activity of sympathetic vasoconstrictor nerves makes the normal contractile tone of arterioles considerably greater than their basal tone. The additional component of vascular tone is called neurogenic tone. When the firing rate of sympathetic vasoconstrictor nerves is increased above normal, arterioles constrict and cause organ blood flow to fall below normal. Conversely, vasodilation and increased organ blood flow can be caused by sympathetic vasoconstrictor nerves if their normal tonic activity level is reduced. Thus, an organ’s blood flow can either be reduced below normal or be increased above normal by changes in the sympathetic vasoconstrictor fiber firing rate.
OTHER NEURAL INFLUENCES
Blood vessels, as a general rule, do not receive innervation from the parasympathetic division of the autonomic nervous system. However, parasympathetic vasodilator nerves, which release acetylcholine, are present in the vessels of the brain and the heart, but their influence on arteriolar tone in these organs appears to be inconsequential. Parasympathetic vasodilator nerves are also present in the vessels of the salivary glands, pancreas, and gastric mucosa where they have important influences on secretion and motility. In the external genitalia, they are responsible for the vasodilation of inflow vessels responsible for promoting secretion and erection.
Hormonal Influences on Arterioles
![]() Under normal circumstances, short-term hormonal influences on blood vessels are generally thought to be of minor consequence in comparison to the local metabolic and neural influences. However, it should be emphasized that the understanding of how the cardiovascular system operates in many situations is incomplete. Thus, the hormones discussed in the following sections may play more important roles in cardiovascular regulation than is now appreciated.
Under normal circumstances, short-term hormonal influences on blood vessels are generally thought to be of minor consequence in comparison to the local metabolic and neural influences. However, it should be emphasized that the understanding of how the cardiovascular system operates in many situations is incomplete. Thus, the hormones discussed in the following sections may play more important roles in cardiovascular regulation than is now appreciated.
CIRCULATING CATECHOLAMINES
During activation of the sympathetic nervous system, the adrenal glands release the catecholamines epinephrine and norepinephrine into the bloodstream. Under normal circumstances, the blood levels of these agents are probably not high enough to cause significant cardiovascular effects. However, circulating catecholamines may have cardiovascular effects in situations (such as vigorous exercise or hemorrhagic shock) that involve high activity of the sympathetic nervous system. In general, the cardiovascular effects of high levels of circulating catecholamines parallel the direct effects of sympathetic activation, which have already been discussed; both epinephrine and norepinephrine can activate cardiac β1-adrenergic receptors to increase the heart rate and myocardial contractility and can activate vascular α-receptors to cause vasoconstriction. Recall that in addition to the α1-receptors that mediate vasoconstriction, arterioles in a few organs also possess β2-adrenergic receptors that mediate vasodilation. Because vascular β2-receptors are more sensitive to epinephrine than are vascular α1-receptors, moderately elevated levels of circulating epinephrine can cause vasodilation, whereas higher levels cause α1-receptor-mediated vasoconstriction. Vascular β2-receptors are not innervated and therefore are not activated by norepinephrine, released from sympathetic vasoconstrictor nerves. The physiological importance of these vascular β2-receptors is unclear because adrenal epinephrine release occurs during periods of increased sympathetic activity when arterioles would simultaneously be undergoing direct neurogenic vasoconstriction. Again, under normal circumstances, circulating catecholamines are not an important factor in cardiovascular regulation.
VASOPRESSIN
![]() This polypeptide hormone, also known as antidiuretic hormone (or ADH), plays an important role in extracellular fluid homeostasis and is released into the bloodstream from the posterior pituitary gland in response to low blood volume and/or high extracellular fluid osmolarity. Vasopressin acts on collecting ducts in the kidneys to decrease renal excretion of water. Its role in body fluid balance has some very important indirect influences on cardiovascular function, which is discussed in more detail in Chapter 9. Vasopressin, however, is also a potent arteriolar vasoconstrictor. Although it is not thought to be significantly involved in normal vascular control, direct vascular constriction from abnormally high levels of vasopressin may be important in the response to certain disturbances such as severe blood loss through hemorrhage.
This polypeptide hormone, also known as antidiuretic hormone (or ADH), plays an important role in extracellular fluid homeostasis and is released into the bloodstream from the posterior pituitary gland in response to low blood volume and/or high extracellular fluid osmolarity. Vasopressin acts on collecting ducts in the kidneys to decrease renal excretion of water. Its role in body fluid balance has some very important indirect influences on cardiovascular function, which is discussed in more detail in Chapter 9. Vasopressin, however, is also a potent arteriolar vasoconstrictor. Although it is not thought to be significantly involved in normal vascular control, direct vascular constriction from abnormally high levels of vasopressin may be important in the response to certain disturbances such as severe blood loss through hemorrhage.
ANGIOTENSIN II
![]() Angiotensin II is a circulating polypeptide that regulates aldosterone release from the adrenal cortex as part of the system for controlling body’s sodium balance. This system, discussed in greater detail in Chapter 9, is very important in blood volume regulation. Angiotensin II is also a very potent vasoconstrictor agent. Although it should not be viewed as a normal regulator of arteriolar tone, direct vasoconstriction from angiotensin II seems to be an important component of the general cardiovascular response to severe blood loss. There is also strong evidence suggesting that direct vascular actions of angiotensin II may be involved in intrarenal mechanisms for controlling kidney function. In addition, angiotensin II may be partially responsible for the abnormal vasoconstriction that accompanies many forms of hypertension. Again, it should be emphasized that knowledge of many pathological situations—including hypertension–is incomplete. These situations may well involve vascular influences that are not yet recognized.
Angiotensin II is a circulating polypeptide that regulates aldosterone release from the adrenal cortex as part of the system for controlling body’s sodium balance. This system, discussed in greater detail in Chapter 9, is very important in blood volume regulation. Angiotensin II is also a very potent vasoconstrictor agent. Although it should not be viewed as a normal regulator of arteriolar tone, direct vasoconstriction from angiotensin II seems to be an important component of the general cardiovascular response to severe blood loss. There is also strong evidence suggesting that direct vascular actions of angiotensin II may be involved in intrarenal mechanisms for controlling kidney function. In addition, angiotensin II may be partially responsible for the abnormal vasoconstriction that accompanies many forms of hypertension. Again, it should be emphasized that knowledge of many pathological situations—including hypertension–is incomplete. These situations may well involve vascular influences that are not yet recognized.
CONTROL OF VENOUS TONE
Before considering the details of the control of venous tone, recall that venules and veins play a much different role in the cardiovascular system than do arterioles. Arterioles are the inflow valves that control the rate of nutritive blood flow through organs and individual regions within them. Appropriately, arterioles are usually strongly influenced by the current local metabolic needs of the region in which they reside, whereas veins are not. Veins do, however, collectively regulate the distribution of available blood volume between the peripheral and central venous compartments. Recall that central blood volume (and therefore pressure) has a marked influence on stroke volume and cardiac output. Consequently, when one considers what peripheral veins are doing, one should be thinking primarily about what the effects will be on central venous pressure and cardiac output.
![]()
![]() Veins contain the vascular smooth muscle that is influenced by many things that influence the vascular smooth muscle of arterioles. Constriction of the veins (venoconstriction) is largely mediated through activity of the sympathetic nerves that innervate them. As in arterioles, these sympathetic nerves release norepinephrine, which interacts with α1-receptors and produces an increase in venous tone and a decrease in vessel diameter. There are, however, several functionally important differences between veins and arterioles. Compared with arterioles, veins normally have little basal tone. Thus, veins are normally in a dilated state. One important consequence of the lack of basal venous tone is that vasodilator metabolites that may accumulate in the tissue have little effect on veins.
Veins contain the vascular smooth muscle that is influenced by many things that influence the vascular smooth muscle of arterioles. Constriction of the veins (venoconstriction) is largely mediated through activity of the sympathetic nerves that innervate them. As in arterioles, these sympathetic nerves release norepinephrine, which interacts with α1-receptors and produces an increase in venous tone and a decrease in vessel diameter. There are, however, several functionally important differences between veins and arterioles. Compared with arterioles, veins normally have little basal tone. Thus, veins are normally in a dilated state. One important consequence of the lack of basal venous tone is that vasodilator metabolites that may accumulate in the tissue have little effect on veins.
Because of their thin walls, veins are much more susceptible to physical influences than are arterioles. The large effect of internal venous pressure on venous diameter was discussed in Chapter 6 and is evident in the pooling of blood in the veins of the lower extremities that occurs during prolonged standing (as discussed further in Chapter 10).
Often external compressional forces are an important determinant of venous volume. This is especially true of veins in the skeletal muscle. Very high pressures are developed inside skeletal muscle tissue during contraction and cause venous vessels to collapse. Because veins and venules have one-way valves, the blood displaced from veins during skeletal muscle contraction is forced in the forward direction toward the right side of the heart. In fact, rhythmic skeletal muscle contractions may produce a considerable pumping action, often called the skeletal muscle pump, which helps return blood to the heart during exercise.
SUMMARY OF PRIMARY VASCULAR CONTROL MECHANISMS
As is apparent from the previous discussion, vessels are subject to a wide variety of influences, and special influences and/or situations often apply to particular organs. Certain general factors, however, dominate the primary control of the peripheral vasculature when it is viewed from the standpoint of overall cardiovascular system function; these influences are summarized in Figure 7–5. Basal tone, local metabolic vasodilator factors, and sympathetic vasoconstrictor nerves acting through α1-receptors are the major factors controlling arteriolar tone and therefore the blood flow rate through peripheral organs. Sympathetic vasoconstrictor nerves, internal pressure, and external compressional forces are the most important influences on venous diameter and therefore on peripheral–central distribution of blood volume.
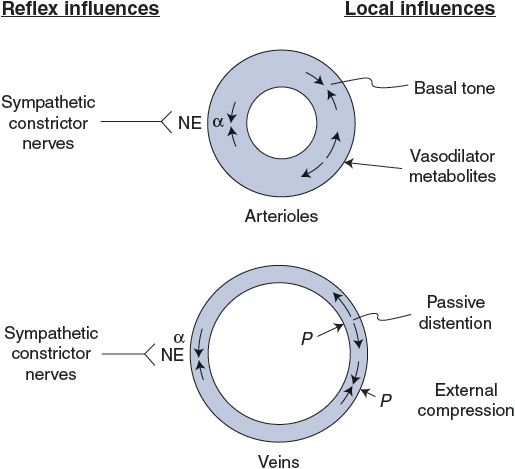
Figure 7–5. Primary influences on arterioles and veins. NE, norepinephrine; α, alpha-adrenergic receptor; P, pressure.
![]()
![]()
![]() As evident in the remaining sections of this chapter, many details of vascular control vary from organs to organs. However, with regard to flow control, most organs can be placed somewhere in a spectrum that ranges from almost total dominance by local metabolic mechanisms to almost total dominance by sympathetic vasoconstrictor nerves.
As evident in the remaining sections of this chapter, many details of vascular control vary from organs to organs. However, with regard to flow control, most organs can be placed somewhere in a spectrum that ranges from almost total dominance by local metabolic mechanisms to almost total dominance by sympathetic vasoconstrictor nerves.
The flow in organs such as the brain, heart muscle, and skeletal muscle is very strongly controlled by local metabolic control, whereas the flow in the kidneys, skin, and splanchnic organs is very strongly controlled by sympathetic nerve activity. Consequently, some organs are automatically forced to participate in overall cardiovascular reflex responses to a greater extent than are other organs. The overall plan seems to be that, in cardiovascular emergency, flow to the brain and heart will be preserved at the expense of everything else if need be.
VASCULAR CONTROL IN SPECIFIC ORGANS
The general types of vascular influences outlined previously in this chapter have different relative importance in different organs. In the following sections, we consider how blood flow control differs between some major organs. Such differences obviously influence what determines the blood flow through the particular organ in question. But it is well to keep in perspective that all organs are part of the overall, hydraulically interconnected cardiovascular system. What happens in any single organ ultimately has ramifications throughout the entire system. In the following summary of flow control in specific organs, we attempt to address both local and global issues by listing the important and sometimes unique factors that control flow in major organs or organ systems.
Coronary Blood Flow
1. The major right and left coronary arteries that serve the heart tissue are the first vessels to branch off the aorta. Thus, the driving force for myocardial blood flow is the systemic arterial pressure, just as it is for other systemic organs. Most of the blood that flows through the myocardial tissue returns to the right atrium by way of a large cardiac vein called the coronary sinus.
2. ![]() Coronary blood flow is controlled primarily by local metabolic mechanisms. It responds rapidly and accurately to changes in myocardial oxygen consumption. In a resting individual, the myocardium extracts 70% to 75% of the oxygen in the blood that passes through it. Because of this high extraction rate, coronary sinus blood normally has a lower oxygen content than blood at any other place in the cardiovascular system.
Coronary blood flow is controlled primarily by local metabolic mechanisms. It responds rapidly and accurately to changes in myocardial oxygen consumption. In a resting individual, the myocardium extracts 70% to 75% of the oxygen in the blood that passes through it. Because of this high extraction rate, coronary sinus blood normally has a lower oxygen content than blood at any other place in the cardiovascular system.
3. Because myocardial oxygen extraction cannot increase significantly from its high resting value, increases in myocardial oxygen consumption must be accompanied by appropriate increases in coronary blood flow.
4. The issue of which metabolic vasodilator factors play the dominant role in modulating the tone of coronary arterioles is unresolved at present. Many suspect that adenosine, released from myocardial muscle cells in response to increased metabolic rate, may be an important local coronary metabolic vasodilator influence. Regardless of the specific details, myocardial oxygen consumption is the most important influence on coronary blood flow.
5. Large forces and/or pressures are generated within the myocardial tissue during cardiac muscle contraction. Such intramyocardial forces press on the outside of coronary vessels and cause them to collapse during systole. Because of this systolic compression and the associated collapse of coronary vessels, coronary vascular resistance is greatly increased during systole. The result, at least for much of the left ventricular myocardium, is that coronary flow is lower during systole than during diastole, even though systemic arterial pressure (ie, coronary perfusion pressure) is highest during systole. This is illustrated in the left coronary artery flow trace shown in Figure 7–6. Systolic compression has much less effect on flow through the right ventricular myocardium, as is evident from the right coronary artery flow trace in Figure 7–6. This is because the peak systolic intraventricular pressure is much lower for the right heart than for the left heart, and the systolic compressional forces in the right ventricular wall are correspondingly less than those in the left ventricular wall.
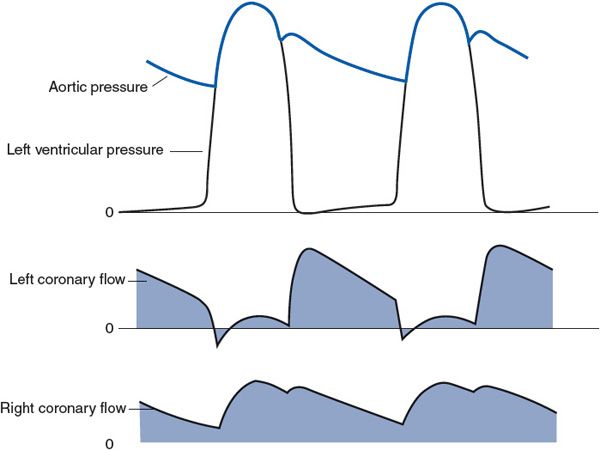
Figure 7–6. Phasic flows in the left and right coronary arteries in relation to aortic and left ventricular pressures.
6. Systolic compressional forces on coronary vessels are greater in the endocardial (inside) layers of the left ventricular wall than in the epicardial layers.4 Thus, the flow to the endocardial layers of the left ventricle is impeded more than the flow to the epicardial layers by systolic compression. Normally, the endocardial region of the myocardium can make up for the lack of flow during systole by a high flow in the diastolic interval. However, when coronary blood flow is limited—for example, by coronary disease and stenosis—the endocardial layers of the left ventricle are often the first regions of the heart to have difficulty maintaining a flow sufficient for their metabolic needs. Myocardial infarcts (areas of tissue killed by lack of blood flow) occur most frequently in the endocardial layers of the left ventricle.
7. Coronary arterioles are densely innervated with sympathetic vasoconstrictor fibers, yet when the activity of the sympathetic nervous system increases, the coronary arterioles normally vasodilate rather than vasoconstrict. This is because an increase in sympathetic tone increases myocardial oxygen consumption by increasing the heart rate and contractility. The increased local metabolic vasodilator influence apparently outweighs the concurrent vasoconstrictor influence of an increase in the activity of sympathetic vasoconstrictor fibers that terminate on coronary arterioles. It has been experimentally demonstrated that a given increase in cardiac sympathetic nerve activity causes a greater increase in coronary blood flow after the direct vasoconstrictor influence of sympathetic nerves on coronary vessels has been eliminated with α-receptor-blocking agents. However, sympathetic vasoconstrictor nerves do not appear to influence coronary flow enough to affect the mechanical performance of normal hearts. Whether these coronary vasoconstrictor fibers might be functionally important in certain pathological situations is still an open question.
Skeletal Muscle Blood Flow
1. Because of the large mass of the skeletal muscle, blood flow through it is an important factor in overall cardiovascular hemodynamics. Collectively, the skeletal muscles constitute 40% to 45% of body weight—more than any other single body organ. Even at rest, approximately 15% of the cardiac output goes to skeletal muscle, and during strenuous exercise, the skeletal muscle may receive more than 80% of the cardiac output.
2. Resting skeletal muscle has a high level of intrinsic vascular tone. Because of this high tone of the smooth muscle in resistance vessels of resting skeletal muscles, the blood flow per gram of tissue is quite low when compared with that of other organs such as the kidneys. However, resting skeletal muscle blood flow is still substantially above that required to sustain its metabolic needs. Resting skeletal muscles normally extract only 25% to 30% of the oxygen delivered to them in arterial blood. Thus, changes in the activity of sympathetic vasoconstrictor fibers can reduce resting muscle blood flow without compromising resting tissue metabolic processes.
3. ![]() Local metabolic control of arteriolar tone is the most important influence on blood flow through exercising muscle. A particularly important characteristic of skeletal muscle is its very wide range of metabolic rates. During heavy exercise, the oxygen consumption rate of and oxygen extraction by skeletal muscle tissue can reach the high values typical of the myocardium. In most respects, the factors that control blood flow to exercising muscle are similar to those that control coronary blood flow. Local metabolic control of arteriolar tone is very strong in exercising skeletal muscle, and muscle oxygen consumption is the most important determinant of its blood flow. Blood flow in the skeletal muscle can increase 20-fold during a bout of strenuous exercise.
Local metabolic control of arteriolar tone is the most important influence on blood flow through exercising muscle. A particularly important characteristic of skeletal muscle is its very wide range of metabolic rates. During heavy exercise, the oxygen consumption rate of and oxygen extraction by skeletal muscle tissue can reach the high values typical of the myocardium. In most respects, the factors that control blood flow to exercising muscle are similar to those that control coronary blood flow. Local metabolic control of arteriolar tone is very strong in exercising skeletal muscle, and muscle oxygen consumption is the most important determinant of its blood flow. Blood flow in the skeletal muscle can increase 20-fold during a bout of strenuous exercise.
4.![]() Alterations in sympathetic neural activity can alter nonexercising skeletal muscle blood flow. For example, maximum sympathetic discharge rates can decrease blood flow in a resting muscle to less than one-fourth its normal value, and conversely, if all neurogenic tone is removed, resting skeletal muscle blood flow may double. This is a modest increase in flow compared with what can occur in an exercising skeletal muscle. Nonetheless, because of the large mass of tissue involved, changes in the vascular resistance of resting skeletal muscle brought about by changes in sympathetic activity are very important in the overall reflex regulation of arterial pressure.
Alterations in sympathetic neural activity can alter nonexercising skeletal muscle blood flow. For example, maximum sympathetic discharge rates can decrease blood flow in a resting muscle to less than one-fourth its normal value, and conversely, if all neurogenic tone is removed, resting skeletal muscle blood flow may double. This is a modest increase in flow compared with what can occur in an exercising skeletal muscle. Nonetheless, because of the large mass of tissue involved, changes in the vascular resistance of resting skeletal muscle brought about by changes in sympathetic activity are very important in the overall reflex regulation of arterial pressure.
5. Alterations in sympathetic neural activity can influence exercising skeletal muscle blood flow. As will be discussed in Chapter 10, the cardiovascular response to muscle exercise involves a general increase in sympathetic activity. This of course reduces flow to susceptible organs, which include nonexercising muscles. In exercising muscles, the increased sympathetic vasoconstrictor nerve activity is not evident as outright vasoconstriction but does limit the degree of metabolic vasodilation. One important function that this seemingly counterproductive process may serve is that of preventing an excessive reduction in total peripheral resistance during exercise. Indeed, if arterioles in most of the skeletal muscles in the body were allowed to dilate to their maximum capacity simultaneously, total peripheral resistance would be so low that the heart could not possibly supply enough cardiac output to maintain arterial pressure.
6. Rhythmic contractions can increase venous return from exercising skeletal muscle. As in the heart, muscle contraction produces large compressional forces within the tissue, which can collapse vessels and impede blood flow. Strong, sustained (tetanic) skeletal muscle contractions may actually stop muscle blood flow. Approximately 10% of the total blood volume is normally contained within the veins of the skeletal muscle, and during rhythmic exercise, the “skeletal muscle pump” is very effective in displacing blood from skeletal muscle veins. Valves in the veins prevent reverse flow back into the muscles. Blood displaced from the skeletal muscle into the central venous pool is an important factor in the hemodynamics of strenuous whole body exercise.
7. Veins in skeletal muscle can constrict in response to increased sympathetic activity. However, veins in the skeletal muscle are rather sparsely innervated with sympathetic vasoconstrictor fibers, and the rather small volume of blood that can be mobilized from the skeletal muscle by sympathetic nerve activation is probably not of much significance to total body hemodynamics. This is in sharp contrast to the large displacement of blood from exercising muscle by the muscle pump mechanism. (This is discussed in more detail when postural reflexes are considered in Chapter 10.)
Cerebral Blood Flow
1. Interruption of cerebral blood flow for more than a few seconds leads to unconsciousness and to brain damage within a very short period. One rule of overall cardiovascular system function is that, in all situations, measures are taken that are appropriate to preserve adequate blood flow to the brain. This is normally accomplished by very rapid reflex adjustments in cardiac output and total peripheral resistance designed to keep mean arterial pressure constant (discussed in more detail in Chapters 9 and 10).
2.![]() Cerebral blood flow is regulated almost entirely by local mechanisms. The brain as a whole has a nearly constant rate of metabolism that, on a per gram basis, is nearly as high as that of myocardial tissue. Flow through the cerebrum is autoregulated very strongly and is little affected by changes in arterial pressure unless it falls below approximately 60 mm Hg. When arterial pressure decreases below 60 mm Hg, brain blood flow decreases proportionately. It is presently unresolved whether metabolic mechanisms or myogenic mechanisms or both are involved in the phenomenon of cerebral autoregulation.
Cerebral blood flow is regulated almost entirely by local mechanisms. The brain as a whole has a nearly constant rate of metabolism that, on a per gram basis, is nearly as high as that of myocardial tissue. Flow through the cerebrum is autoregulated very strongly and is little affected by changes in arterial pressure unless it falls below approximately 60 mm Hg. When arterial pressure decreases below 60 mm Hg, brain blood flow decreases proportionately. It is presently unresolved whether metabolic mechanisms or myogenic mechanisms or both are involved in the phenomenon of cerebral autoregulation.
3. Local changes in cerebral blood flow may be influenced by local metabolic conditions. Presumably because the overall average metabolic rate of brain tissue shows little variation, total brain blood flow is remarkably constant over nearly all situations. The cerebral activity in discrete locations within the brain, however, changes from situation to situation. As a result, blood flow to discrete regions is not constant but closely follows the local neuronal activity. The mechanisms responsible for this strong local control of cerebral blood flow are as yet undefined, but H+, K+, oxygen, and adenosine seem most likely to be involved.
4. Cerebral blood flow decreases whenever arterial blood PCO2 falls below normal. Conversely, cerebral blood flow increases whenever the partial pressure of carbon dioxide (PCO2) is raised above normal in the arterial blood. This is the normal state of affairs in most tissues, but it plays out importantly when it happens in the brain. For example, the dizziness, confusion, and even fainting that can occur when a person hyperventilates (and “blows off” CO2) are a direct result of cerebral vasoconstriction. It appears that cerebral arterioles respond not to changes in Pco2 but to changes in the extracellular H+ concentration (ie, pH) caused by changes in PCO2. Cerebral arterioles also vasodilate whenever the partial pressure of oxygen (PO2) in arterial blood falls significantly below normal values. However, higher-than-normal arterial blood Po2, such as that caused by pure oxygen inhalation, produces little decrease in cerebral blood flow.
5. Sympathetic and parasympathetic neural influences on cerebral blood flow are minimal. Although cerebral vessels receive both sympathetic vasoconstrictor and parasympathetic vasodilator fiber innervation, cerebral blood flow is influenced very little by changes in the activity of either under normal circumstances. Sympathetic vasoconstrictor responses may, however, be important in protecting cerebral vessels from excessive passive distention following large, abrupt increases in arterial pressure.
6. The “blood–brain barrier” refers to the tightly connected vascular endothelial cells that severely restrict transcapillary movement of all polar and many other substances.5 Because of this blood–brain barrier, the extracellular space of the brain represents a special fluid compartment in which the chemical composition is regulated separately from that in the plasma and general body extracellular fluid compartment. The extracellular compartment of the brain encompasses both interstitial fluid and cerebrospinal fluid (CSF), which surrounds the brain and the spinal cord and fills the brain ventricles. The CSF is formed from plasma by selective secretion (not simple filtration) by specialized tissues, the choroid plexes, located within the brain ventricles. These processes regulate the chemical composition of the CSF. The interstitial fluid of the brain takes on the chemical composition of CSF through free diffusional exchange.
The blood–brain barrier serves to protect the cerebral cells from ionic disturbances in the plasma. Also, by exclusion and/or endothelial cell metabolism, it prevents many circulating hormones (and drugs) from influencing the parenchymal cells of the brain and the vascular smooth muscle cells in brain vessels.
7. Although many organs can tolerate some level of edema (the accumulation of excess extracellular fluid), edema in the brain represents a crisis situation. Cerebral edema increases intracranial pressure, which must be promptly relieved to avoid brain damage. Special mechanisms involving various specific ion channels and transporters precisely regulate the transport of solute and water across astrocytes and the endothelial barrier. These mechanisms contribute to normal maintenance of intracellular and extracellular fluid balance.
Splanchnic Blood Flow
1. Because of the high blood flow through and the high blood volume in the splanchnic bed, its vascular control importantly influences overall cardiovascular hemodynamics. A number of abdominal organs, including the gastrointestinal tract, spleen, pancreas, and liver, are collectively supplied with what is called the splanchnic blood flow. Splanchnic blood flow is supplied to these abdominal organs through many arteries, but it all ultimately passes through the liver and returns to the inferior vena cava through the hepatic veins. The organs of the splanchnic region receive approximately 25% of the resting cardiac output and contain more than 20% of the circulating blood volume. Thus, adjustments in either the blood flow or the blood volume of this region have extremely important effects on the cardiovascular system.
2.![]() Sympathetic neural activity plays an important role in vascular control of the splanchnic circulation. Collectively, the splanchnic organs have a relatively high blood flow and extract only 15% to 20% of the oxygen delivered to them in the arterial blood. The arteries and veins of all the organs involved in the splanchnic circulation are richly innervated with sympathetic vasoconstrictor nerves. Maximal activation of sympathetic vasoconstrictor nerves can produce an 80% reduction in flow to the splanchnic region and also cause a large shift of blood from the splanchnic organs to the central venous pool. In humans, a large fraction of the blood mobilized from the splanchnic circulation during periods of sympathetic activation comes from the constriction of veins in the liver. In many other species, the spleen acts as a major reservoir from which blood is mobilized by sympathetically mediated contraction of the smooth muscle located in the outer capsule of the organ.
Sympathetic neural activity plays an important role in vascular control of the splanchnic circulation. Collectively, the splanchnic organs have a relatively high blood flow and extract only 15% to 20% of the oxygen delivered to them in the arterial blood. The arteries and veins of all the organs involved in the splanchnic circulation are richly innervated with sympathetic vasoconstrictor nerves. Maximal activation of sympathetic vasoconstrictor nerves can produce an 80% reduction in flow to the splanchnic region and also cause a large shift of blood from the splanchnic organs to the central venous pool. In humans, a large fraction of the blood mobilized from the splanchnic circulation during periods of sympathetic activation comes from the constriction of veins in the liver. In many other species, the spleen acts as a major reservoir from which blood is mobilized by sympathetically mediated contraction of the smooth muscle located in the outer capsule of the organ.
3.![]() Local metabolic activity associated with gastrointestinal motility, secretion, and absorption is associated with local increases in splanchnic blood flow. There is great diversity of vascular structure and function among individual organs and even regions within organs in the splanchnic region. The mechanisms of vascular control in specific areas of the splanchnic region are not well understood but are likely to be quite varied. Nonetheless, because most splanchnic organs are involved in the digestion and absorption of food from the gastrointestinal tract, overall splanchnic blood flow increases after food ingestion. Parasympathetic neural activity is involved in many of these gastrointestinal functions, so it is indirectly involved in increasing splanchnic blood flow. A large meal can elicit a 30% to 100% increase in splanchnic flow, but individual organs in the splanchnic region probably have higher percentage increases in flow at certain times because they are involved sequentially in the digestion–absorption process.
Local metabolic activity associated with gastrointestinal motility, secretion, and absorption is associated with local increases in splanchnic blood flow. There is great diversity of vascular structure and function among individual organs and even regions within organs in the splanchnic region. The mechanisms of vascular control in specific areas of the splanchnic region are not well understood but are likely to be quite varied. Nonetheless, because most splanchnic organs are involved in the digestion and absorption of food from the gastrointestinal tract, overall splanchnic blood flow increases after food ingestion. Parasympathetic neural activity is involved in many of these gastrointestinal functions, so it is indirectly involved in increasing splanchnic blood flow. A large meal can elicit a 30% to 100% increase in splanchnic flow, but individual organs in the splanchnic region probably have higher percentage increases in flow at certain times because they are involved sequentially in the digestion–absorption process.
Renal Blood Flow
1. Renal blood flow plays a critical role in the kidney’s main long-term job of regulating the body’s water balance and therefore circulating blood volume. However, acute adjustments in renal blood flow also have important short-term hemodynamic consequences. The kidneys normally receive approximately 20% of the cardiac output of a resting individual. This flow can be reduced to practically zero during strong sympathetic activation. Thus, the control of renal blood flow is important to overall cardiovascular function. However, because the kidneys are such small organs, changes in renal blood volume are inconsequential to overall cardiovascular hemodynamics.
2.![]() Renal blood flow is strongly influenced by sympathetic neural stimulation. Alterations in sympathetic neural activity can have marked effects on total renal blood flow by altering the neurogenic tone of renal resistance vessels. In fact, extreme situations involving intense and prolonged sympathetic vasoconstrictor activity (as may accompany severe blood loss) can lead to dramatic reduction in renal blood flow, permanent kidney damage, and renal failure.
Renal blood flow is strongly influenced by sympathetic neural stimulation. Alterations in sympathetic neural activity can have marked effects on total renal blood flow by altering the neurogenic tone of renal resistance vessels. In fact, extreme situations involving intense and prolonged sympathetic vasoconstrictor activity (as may accompany severe blood loss) can lead to dramatic reduction in renal blood flow, permanent kidney damage, and renal failure.
3. Local metabolic mechanism may influence local vascular tone, but physiological roles are not clear. It has long been known that experimentally isolated kidneys (ie, kidneys deprived of their normal sympathetic input) autoregulate their flow quite strongly. The mechanism responsible for this phenomenon has not been definitely established, but myogenic, tissue pressure, and metabolic hypotheses have been advanced. The real question is what purpose such a strong local mechanism plays in the intact organism where it seems to be largely overridden by reflex mechanisms. In an intact individual, renal blood flow is not constant but is highly variable, depending on the prevailing level of sympathetic vasoconstrictor nerve activity.
The mechanisms responsible for the intrinsic regulation of renal blood flow and kidney function have not been established. Although studies suggest that prostaglandins and some intrarenal renin–angiotensin system may be involved, the whole issue of local renal vascular control remains quite obscure. Renal function is itself of paramount importance to overall cardiovascular function, as described in Chapter 9.
Cutaneous Blood Flow
1. The physiological role of skin blood flow is to help regulate body temperature. The metabolic activity of body cells produces heat, which must be lost in order for the body temperature to remain constant. The skin is the primary site of exchange of body heat with the external environment. Alterations in cutaneous blood flow in response to various metabolic states and environmental conditions provide the primary mechanism responsible for temperature homeostasis. (Other mechanisms such as shivering, sweating, and panting also participate in body temperature regulation under more extreme conditions.)
2. Decreases in body temperature decrease skin blood flow and vice versa. Cutaneous blood flow, which is approximately 6% of resting cardiac output, can decrease to about one-twentieth of its normal value when heat is to be retained (eg, in a cold environment, during the development stages of a fever). In contrast, cutaneous blood flow can increase up to seven times its normal value when heat is to be lost (eg, in a hot environment, accompanying a high metabolic rate, after a fever “breaks”).
3. Structural adaptations of the cutaneous vascular beds promote heat loss or heat conservation. The anatomic interconnections between microvessels in the skin are highly specialized and extremely complex. An extensive system of interconnected veins called the venous plexus normally contains the largest fraction of cutaneous blood volume, which, in individuals with lightly pigmented skin, gives the skin a reddish hue. To a large extent, heat transfer from the blood takes place across the large surface area of the venous plexus. The venous plexus is richly innervated with sympathetic vasoconstrictor nerves. When these fibers are activated, blood is displaced from the venous plexus, and this helps reduce heat loss and also lightens the skin color. Because the skin is one of the largest body organs, venous constriction can shift a considerable amount of blood into the central venous pool.
4.![]() Reflex sympathetic neural activity has important but complex influences on skin blood flow. Cutaneous resistance vessels are richly innervated with sympathetic vasoconstrictor nerves, and because these fibers have a normal tonic activity, cutaneous resistance vessels normally have a high degree of neurogenic tone. When body temperature rises above normal, skin blood flow is increased by reflex mechanisms. In certain areas (such as the hands, ears, and nose), vasodilation appears to result entirely from the withdrawal of sympathetic vasoconstrictor tone. In other areas (such as the forearm, forehead, chin, neck, and chest), the cutaneous vasodilation that occurs with body heating greatly exceeds that which occurs with just the removal of sympathetic vasoconstrictor tone. This “active” vasodilation is closely linked to the onset of sweating in these areas. The sweat glands in human cutaneous tissue involved in thermoregulation are innervated by cholinergic sympathetic fibers that release acetylcholine. Activation of these nerves elicits sweating and an associated marked cutaneous vasodilation. The exact mechanism for this sweating-related cutaneous vasodilation remains unclear because it is not abolished by agents that block acetylcholine’s vascular effects. It has long been thought that it was caused by local bradykinin formation secondary to the process of sweat gland activation. Newer evidence suggests that the cholinergic sympathetic nerves to sweat glands may release not only acetylcholine but also other vasodilator cotransmitters. Although these special sympathetic nerves are very important to temperature regulation, they do not participate in the normal, moment-to-moment regulation of the cardiovascular system.
Reflex sympathetic neural activity has important but complex influences on skin blood flow. Cutaneous resistance vessels are richly innervated with sympathetic vasoconstrictor nerves, and because these fibers have a normal tonic activity, cutaneous resistance vessels normally have a high degree of neurogenic tone. When body temperature rises above normal, skin blood flow is increased by reflex mechanisms. In certain areas (such as the hands, ears, and nose), vasodilation appears to result entirely from the withdrawal of sympathetic vasoconstrictor tone. In other areas (such as the forearm, forehead, chin, neck, and chest), the cutaneous vasodilation that occurs with body heating greatly exceeds that which occurs with just the removal of sympathetic vasoconstrictor tone. This “active” vasodilation is closely linked to the onset of sweating in these areas. The sweat glands in human cutaneous tissue involved in thermoregulation are innervated by cholinergic sympathetic fibers that release acetylcholine. Activation of these nerves elicits sweating and an associated marked cutaneous vasodilation. The exact mechanism for this sweating-related cutaneous vasodilation remains unclear because it is not abolished by agents that block acetylcholine’s vascular effects. It has long been thought that it was caused by local bradykinin formation secondary to the process of sweat gland activation. Newer evidence suggests that the cholinergic sympathetic nerves to sweat glands may release not only acetylcholine but also other vasodilator cotransmitters. Although these special sympathetic nerves are very important to temperature regulation, they do not participate in the normal, moment-to-moment regulation of the cardiovascular system.
5. Cutaneous vessels respond to changes in local skin temperature. In general, local cooling leads to local vasoconstriction and local heating causes local vasodilation. The mechanisms for this are unknown. If the hand is placed in ice water, there is initially a nearly complete cessation of hand blood flow accompanied by intense pain. After some minutes, hand blood flow begins to rise to reach values greatly in excess of the normal value, hand temperature increases, and the pain disappears. This phenomenon is referred to as cold-induced vasodilation. With continued immersion, hand blood flow cycles every few minutes between periods of essentially no flow and periods of hyperemia. The mechanism responsible for cold vasodilation is unknown, but it has been suggested that norepinephrine may lose its ability to constrict vessels when their temperature approaches 0°C. Whatever the mechanism, cold-induced vasodilation apparently serves to protect exposed tissues from cold damage.
6. Cutaneous vessels respond to local damage with observable responses. Tissue damage from burns, ultraviolet radiation, cold injury, caustic chemicals, and mechanical trauma produces reactions in skin blood flow. A classical reaction called the triple response is evoked after vigorously stroking the skin with a blunt point. The first component of the triple response is a red line that develops along the direct path of the abrasion in approximately 15 s. Shortly thereafter, an irregular red flare appears that extends approximately 2 cm on either side of the red line. Finally, after a minute or two, a wheal appears along the line of the injury. The mechanisms involved in the triple response are not well understood, but it seems likely that histamine release from damaged cells is at least partially responsible for the dilation evidenced by the red line and the subsequent edema formation of the wheal. The red flare seems to involve nerves in some sort of a local axon reflex, because it can be evoked immediately after cutaneous nerves are sectioned but not after the peripheral portions of the sectioned nerves degenerate.
Pulmonary Blood Flow
1. Pulmonary blood flow equals cardiac output. Except for very transient adjustments, the rate of blood flow through the lungs is necessarily equal to cardiac output of the left ventricle in all circumstances. When cardiac output to the systemic circulation increases threefold during exercise, for example, pulmonary blood flow must also increase threefold.
2. Pulmonary vascular resistance is about one-seventh of total systemic vascular resistance. Pulmonary vessels do offer some vascular resistance. Although the level of pulmonary vascular resistance does not usually influence the pulmonary flow rate, it is important because it is one of the determinants of pulmonary arterial pressure (ΔP = R). Recall that mean pulmonary arterial pressure is approximately 13 mm Hg, whereas mean systemic arterial pressure is approximately 100 mm Hg. The reason for the difference in pulmonary and systemic arterial pressures is not that the right side of the heart is weaker than the left side of the heart, but rather that pulmonary vascular resistance is inherently much lower than systemic total peripheral resistance. The pulmonary bed has a low resistance because it has relatively large vessels throughout.
3. Pulmonary arteries and arterioles are less muscular and more compliant than systemic arteries and arterioles. When pulmonary arterial pressure increases, the pulmonary arteries and arterioles become larger in diameter. Thus, an increase in pulmonary arterial pressure decreases pulmonary vascular resistance. This phenomenon is important because it tends to limit the increase in pulmonary arterial pressure that occurs with increases in cardiac output.
4. Pulmonary arterioles constrict in response to local alveolar hypoxia. One of the most important and unique active responses in pulmonary vasculature is hypoxic vasoconstriction of pulmonary arterioles in response to low oxygen levels within alveoli. (Note: This is a response to alveolar hypoxia, not to low levels of oxygen in the blood—ie, hypoxemia.) This is exactly opposite to the vasodilation that occurs in systemic arterioles in response to low tissue PO2. The mechanisms that cause this unusual response in pulmonary vessels are unclear but seem to be dependent upon oxygen sensing by the pulmonary arterial smooth muscle cells. Current evidence suggests that local endothelin production or prostaglandin synthesis may be involved in pulmonary hypoxic vasoconstriction. Whatever the mechanism, hypoxic vasoconstriction is essential to efficient lung gas exchange because it diverts blood flow away from areas of the lung that are underventilated. Consequently, the best-ventilated areas of the lung also receive the most blood flow. As a consequence of hypoxic arteriolar vasoconstriction, general hypoxia (such as that encountered at high altitude) causes an increase in pulmonary vascular resistance and pulmonary arterial hypertension.
5. Autonomic nerves play no major role in control of pulmonary vascular activity. Both pulmonary arteries and veins receive sympathetic vasoconstrictor fiber innervation, but reflex influences on pulmonary vessels appear to be much less important than the physical and local hypoxic influences. Pulmonary veins serve a blood reservoir function for the cardiovascular system, and sympathetic vasoconstriction of pulmonary veins may be important in mobilizing this blood during periods of general cardiovascular stress.
6. Low capillary hydrostatic pressure promotes fluid reabsorption and prevents fluid accumulation in pulmonary airways. A consequence of the low mean pulmonary arterial pressure is the low pulmonary capillary hydrostatic pressure of approximately 8 mm Hg (compared with approximately 25 mm Hg in systemic capillaries). Because the plasma oncotic pressure in lung capillaries is near 25 mm Hg, as it is in all capillaries, it is likely that the transcapillary forces in the lungs strongly favor continual fluid reabsorption. This cannot be the complete story, however, because the lungs, like other tissues, continually produce some lymph and some net capillary filtration is required to produce lymphatic fluid. This filtration is possible despite the unusually low pulmonary capillary hydrostatic pressure because pulmonary interstitial fluid has an unusually high protein concentration and thus oncotic pressure.
PERSPECTIVES
As should be evident from the broad overview attempted in this chapter, vascular control is indeed a very complex issue. Our current understanding of many of the factors involved is still quite “fuzzy” at best. For openers, we do not understand how vascular smooth muscle itself works as well as we understand how striated muscles work. If that were not enough, smooth muscle operation seems to be potentially influenced by vastly more chemical and mechanical factors than does that of striated muscle. Because of recent advances in cellular and molecular biology, we are now beginning to understand the intricate multiple molecular steps through which some of these pathways act to influence operation of vascular smooth muscle cells. This, of course, has been a stimulus to the pharmaceutical industry to develop drugs that can block (or enhance) this pathway or that. But knowing the mechanism through which a particular influence acts really does nothing to answer the basic issues a practicing physician must face. For example: Do multiple influences just add or do they interact in complex ways? Is the mix of influences importantly different between organs or even within an organ? Is there some adaptation to an influence so that its effect diminishes over time? How is blood flow controlled in transplanted organs? There is much that we do not understand.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


