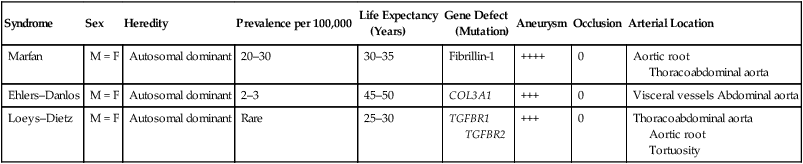
Three connective tissue abnormalities are of particular interest: Marfan syndrome, Ehlers–Danlos syndrome, and Loeys–Dietz syndrome. They are rare diseases, but because they can lead to vascular catastrophes, it is important that vascular surgeons have a basic knowledge about patients who have these syndromes (Table 1). The final diagnosis should be based on molecular genetic testing, in that many signs and symptoms have various degrees of penetrance, making differential diagnosis sometimes difficult. TABLE 1 Comparative Aspects of Connective Tissue Disorders with Vascular Complications
Vascular Complications of Marfan Syndrome, Ehlers–Danlos Syndrome, and Loeys–Dietz Syndrome
Syndrome
Sex
Heredity
Prevalence per 100,000
Life Expectancy
(Years)
Gene Defect
(Mutation)
Aneurysm
Occlusion
Arterial Location
Marfan
M = F
Autosomal dominant
20–30
30–35
Fibrillin-1
++++
0
Aortic root
Thoracoabdominal aorta
Ehlers–Danlos
M = F
Autosomal dominant
2–3
45–50
COL3A1
+++
0
Visceral vessels Abdominal aorta
Loeys–Dietz
M = F
Autosomal dominant
Rare
25–30
TGFBR1
TGFBR2
+++
0
Thoracoabdominal aorta
Aortic root
Tortuosity
Vascular Complications of Marfan Syndrome, Ehlers-Danlos Syndrome, and Loeys-Dietz Syndrome



