Uncommon Primary Malignant Tumors of the Lung
Thomas W. Shields
Philip G. Robinson
Most of the uncommon primary malignant tumors of the lungs are sarcomas, but they also comprise pleomorphic carcinomas, pulmonary blastomas, carcinosarcomas, primary pulmonary lymphomas (PPLs), and various other malignancies, including the rare primary malignant melanoma, primary teratoma, and malignant ependymoma (Table 125-1). In an 11-year review (1980–1990) of 80 rare pulmonary neoplasms seen at the Mayo Clinic, Miller and Allen182 reported that 41% were non-Hodgkin’s lymphomas, 20% were carcinosarcomas, 15% were mucoepidermoid carcinomas (these tumors are discussed in Chapter 123), and 18% were sarcomas; the remainder were either malignant melanomas or blastomas. The patients had a median age of 60 years. In contrast, Hancock and colleagues96 described the distribution of lung tumors in children. They added nine cases to the literature and summarized a total of 383 cases. Most of the tumors (76%) were malignant. Of these, 40.5% were bronchial “adenomas” and 16.8% were bronchial carcinomas. The remaining malignant tumors were pulmonary blastomas (15.5%), lymphomas or plasmacytomas (2.4%), and malignant teratomas (1%). Benign tumors made up 24% of the cases, with the majority of these being inflammatory pseudotumors.
Soft Tissue Sarcomas
Primitive mesenchymal cells are present in every organ of the human body. In the lung, tumors of mesenchymal origin may arise from the stromal elements of the bronchial or vascular wall or from the interstices of lung parenchyma. These tumors usually expand toward the lung parenchyma; occasionally they extend into the lumen of a bronchus. Only rarely do they invade and break through the bronchial epithelium. As a result, these tumors do not exfoliate cells, and diagnosis by cytologic examination of expectorations or of tracheobronchial washings is uncommon. Grossly, the tumor usually appears as a well-circumscribed and encapsulated mass in the lung parenchyma (Fig. 125-1). They generally spread by local invasion. Peripheral lesions may invade the adjacent pleura and chest wall; only rarely do they cavitate. They can metastasize by way of the circulation and rarely by lymphatic invasion. As Watson and Anlyan264 have noted, metastases to distant organs are usually late manifestations of the disease process. Microscopically, these tumors present a wide range of cellular differentiation.
Primary pulmonary sarcomas occur at almost any age, with equal frequency in males and females. Fadhli and colleagues70 report an age range of 4 to 83 years. The tumors occur with equal frequency in either lung. Many patients are asymptomatic, and lesions are detected only on a routine radiograph of the chest. Symptomatic patients most commonly experience chest pain, cough, dyspnea, and hemoptysis. Fever, fatigue, anorexia, and weight loss usually are late manifestations. On radiographs of the chest, the tumor usually appears as a sharply demarcated mass density within the lung substance at the hilum or in the lung periphery. The lesions are usually solitary. Martini and associates173 report that these tumors vary in diameter from 1 to 15 cm or more, with an average diameter of 6 to 7 cm. Peripheral tumors invading the chest wall may be associated with varying degrees of pleural effusion. Tumors obstructing a bronchus (∼15%) may result in distal parenchymal changes (Fig. 125-2).
Keel and coworkers129 have reported a study of 26 primary pulmonary sarcomas. The tumors were distributed as follows: seven malignant fibrous histiocytomas (27%), six synovial sarcomas (23%), three malignant peripheral nerve sheath tumors (12%), three leiomyosarcomas (12%), two angiosarcomas (8%), two intimal sarcomas (8%), two fibrosarcomas (8%), and one epithelioid hemangioendothelioma (4%). The patients ranged in age from 18 to 75 years, with a mean age of 48 years. The tumors ranged in size from 0.9 cm to filling the entire hemithorax. The tumors were distributed almost equally between the two lungs, with one being bilateral, two in the pulmonary artery, and one in a location that was not stated. In a follow-up study of 23 of these patients by Bacha and colleagues,8 it was recorded that 3 patients were unresectable; 13 patients were treated by surgical resections that included lobectomies, bilobectomies, sleeve resections, a carinal resection, and a chest wall resection; 4 patients had radical pneumonectomies; and 3 patients with vascular or cardiac invasion underwent extended resection with the use of cardiopulmonary bypass; 11 patients received postoperative chemotherapy and 8 had radiation therapy. In the follow-up on 22 of the patients, which ranged from 2 to 183 months with a mean of 48 months, 14 patients were free of disease, 6 died of disease, 1 died of surgical complications, and 2 were alive with disease. The size and grade did not correlate with the survival
but the completeness of the resection did. Thus these authors concluded that patients with pulmonary sarcomas may have an acceptable survival rate if the resection is complete.
but the completeness of the resection did. Thus these authors concluded that patients with pulmonary sarcomas may have an acceptable survival rate if the resection is complete.
Table 125-1 Less Common Malignant Tumors of the Lung | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
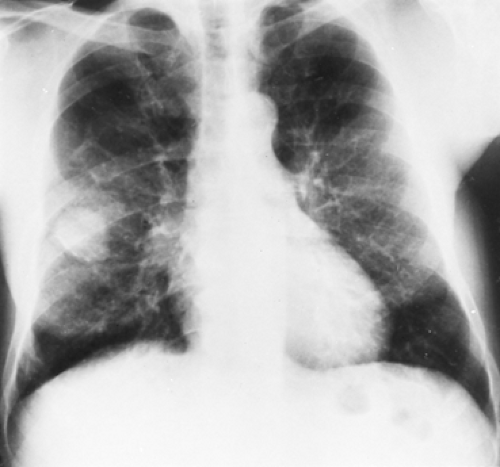 Figure 125-1. Postanterior chest radiograph reveals recurrent fibrosarcoma of the right lung after previous wedge resection of a “fibromatous” tumor from the middle lobe. |
 Figure 125-2. Malignant endobronchial sarcoma. |
In another study of primary lung sarcomas, Magne and colleagues166 reported on nine patients: five women and four men, ranging in age from 35 to 73 years with a median of 63 years. They complained of chest pain (four patients) and dyspnea (four patients). All underwent surgery, with three having incomplete resections. The tumors ranged in size from 2 to 15 cm; they consisted of four malignant fibrous histiocytomas, three leiomyosarcomas, one fibrosarcoma, and one myxoid liposarcoma. Five patients received adjuvant chemotherapy (containing anthracyclin and ifosfamide) and three received adjuvant radiation therapy. The median overall survival was 36 months. In the three patients with incomplete resections, the median survival was 47 months. Two patients underwent a second procedure for the management of recurrence, and each had a satisfactory long-term survival (58 and 83 months, respectively). This good long-term survival for patients with resected recurrence stresses the importance of close follow-up for these patients. The important prognostic factors are low grade of the malignancy, small size of the tumor, and initial complete resection.
Dail53 categorized the soft tissue sarcomas arising within the lung into three groups: (a) parenchymal and bronchial–endobronchial sarcomas, (b) sarcomas of large vessel origin, and (c) sarcomas of small vessel origin.
Parenchymal and Bronchial– Endobronchial Sarcomas
Fibrosarcoma, leiomyosarcoma, rhabdomyosarcoma, neurogenic sarcoma, chondrosarcoma, osteosarcoma, synovial sarcoma, malignant mesenchymoma, liposarcoma, and malignant fibrous histiocytoma are included in this category. These sarcomas may occur in either an endobronchial or a parenchymal location; however, the occurrence of any of these sarcomas as primary lesions in the lung is rare. Table 125-2 is a list of the distribution of some of these lesions in adults and children in the series reported by Hartman and Shochat,102 McCormack and Martini,177 Nascimento,195 and Janssen121 and their associates.
Table 125-2 Distribution of Soft Tissue Sarcomas | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Pulmonary Fibrosarcoma
Combining the patients in the series of Guccion and Rosen91 and Nascimento and colleagues195 yields a total of 22 cases of primary pulmonary fibrosarcomas. The patients ranged in age from 23 to 69 years, with an average age of 47 years. There were 16 (72%) men and 6 (27%) women. This bias might exist because one of the studies is from the Armed Forces Institute of Pathology. These fibrosarcomas may occur in either an endobronchial or a parenchymal location. The endobronchial tumors were almost always symptomatic. The symptoms ranged from none to chest pain, cough, hemoptysis, and shortness of breath. McLigeyo and associates180 reported a pulmonary fibrosarcoma in a 50-year-old woman who presented with hypoglycemia and hypertrophic pulmonary osteoarthropathy. Most patients were treated mainly by surgical excision. A few patients received radiation therapy and chemotherapy. In Guccion and Rosen’s91 series, several patients were lost to follow-up, but the majority of the followed patients died of their disease. Those patients with endobronchial lesions seemed to survive longer. In the nine cases of Nascimento and colleagues,195 seven died from their disease within 3 months to 18 years after treatment and two were alive at 7 and 18 years following treatment. The patient who survived 18 years had an endobronchial lesion. The one who survived 7 years had a parenchymal tumor.
Pulmonary fibrosarcoma is rare in children. Kuhnen and associates148 described a pulmonary spindle-cell tumor in a newborn that they believed to be an infantile fibrosarcoma. Prognosis is thought to be excellent after resection of such a lesion. Picard and colleagues207 resected a bronchial fibrosarcoma by a sleeve resection of the left upper lobe in an 8-year-old boy; the patient was alive and well 2 years after resection. Goldthorn and colleagues87 described a cavitating fibrosarcoma in an 11-year-old girl who underwent a resection followed by chemotherapy. She had a 36-month disease-free survival. Picard and associates,207 in their review of bronchopulmonary fibrosarcomas, noted that at least 28 cases have been recorded in children; 6 were newborns and the others ranged in age from 1 month to 19 years. Each gender was approximately equally affected, as were the two lungs. According to Hancock and colleagues,96 these tumors account for 9.6% of the cases of pediatric lung tumors. In children, the tumor is low-grade and the prognosis is relatively satisfactory with an early survival rate of approximately 78%. Complete surgical resection is the procedure of choice. The roles of radiation therapy and chemotherapy are unknown.
Pulmonary Leiomyosarcoma
Guccion and Rosen91 as well as Nascimento195 and Moran186 and their colleagues have reported a total of 41 cases of pulmonary leiomyosarcomas. These patients ranged in age from a newborn to 91 years, with an average of 51 years. There were 29 men and 12 women, for a gender ratio of 2.4:1.0. The symptoms ranged from none to cough, chest pain, dyspnea, and hemoptysis. The chest radiographs usually showed a solitary homogeneous density with sharply lobulated borders. Cavitation was observed in some of the leiomyosarcomas, as noted by Lillo-Gil and associates.161 The tumors were randomly distributed in all the lobes of both lungs. On gross examination, the leiomyosarcoma may be in an endobronchial, parenchymal, or subpleural location and is usually well circumscribed, firm, and gray-white. Areas of necrosis or hemorrhage may exist. Microscopically, the tumor comprises spindle cells with broad fascicles that intersect at right angles. Distant metastases may occur to the lung and not infrequently the adrenal glands are involved.
Moran and colleagues186 divided the pulmonary leiomyosarcomas into low-grade malignancy, intermediate-grade malignancy, and high-grade malignancy. The low-grade neoplasms have a well-developed fascicular pattern. The cells have “cigar-shaped” nuclei without much atypia, eosinophilic cytoplasm, and 1 to 3 mitoses per 10 high power fields (HPFs). The intermediate-grade tumors have a fairly well-preserved fascicular pattern but are more cellular. The cells have more atypical hyperchromatic nuclei. Mitoses are increased to 3 to 8 per 10 HPFs. Areas of hemorrhage and necrosis are not apparent. High-grade tumors show a more solid cellular proliferation with
less of a fascicular pattern. The cells are markedly pleomorphic with large hyperchromatic nuclei and prominent nucleoli. Mitoses are increased to 8 to 12 per 10 HPFs, and areas of hemorrhage and necrosis are present. Immunostaining shows that most of the tumors (75%) stain for smooth muscle actin.
less of a fascicular pattern. The cells are markedly pleomorphic with large hyperchromatic nuclei and prominent nucleoli. Mitoses are increased to 8 to 12 per 10 HPFs, and areas of hemorrhage and necrosis are present. Immunostaining shows that most of the tumors (75%) stain for smooth muscle actin.
Treatment is surgical removal of the tumor. Shimizu and associates239 resected a primary pulmonary leiomyosarcoma and a hepatic metastasis in a single-stage procedure. Whether such aggressive intervention is appropriate is questionable. A small number (three) of extensive leiomyosarcomas of the lung that involved the heart or great vessels have been excised completely with support of cardiopulmonary bypass during the resection by Weibe and associates.265 These patients tolerated the procedure and have done well postoperatively. The three patients were alive and well for 15, 61, and 216 months respectively. Other sarcomas of the lung likewise appear to do well with this radical approach when the patient is selected with special care and the resection of the tumor is complete. However, when a pneumonectomy is required, the mortality may be as high as 15%. The results obtained in this series in in contrast to the dismal results when cardiopulmonary bypass is used to support the resection of extensive non-small-cell lung cancer associated with lymph node metastases. Three such patients were recorded in the same report and the survival was only 4, 6, and 26 months in these three patients. Byrne26 and Park203 and their colleagues, among others, have reported variable results with this approach. Proper selection of patients is a most important key for success.
As a rule the prognosis is poor, with the majority of the patients dying of the disease, although a few patients have survived for 15 to 20 years. Muscolino and associates190 reported successful resections and long-term survivals (7 years) in two patients with low-grade bronchial leiomyosarcoma. This report supports the conclusion of Moran and colleagues186 that the grade of the tumor was one of the more important factors in determining the patient’s prognosis.
Pulmonary Rhabdomyosarcoma
Rhabdomyosarcomas are malignant tumors of skeletal muscle and may occur in various age groups and in either sex. The majority of the cases occur in infants and children from the ages of 1.5 years to 14 years, but Przygodzki and associates214 have described three cases in men ranging in age from 57 to 78 years. Comin and colleagues43 reported a primary pulmonary rhabdomyosarcoma in a 62-year-old patient. The symptoms depend on whether the lesion is endobronchial or parenchymal. According to d’Agostino and colleagues,52 many of the rhabdomyosarcomas that arise in children are associated with cystic adenomatoid malformations. The chest radiographs usually show a mass that may contain cysts. The computed tomographic (CT) scan shows a soft tissue mass that may be cystic. Pathologically, the tumors have reddish gray surfaces with hemorrhagic and necrotic areas. On microscopic examination, the cells may be arranged in fascicles or may be randomly organized. The nuclei are hyperchromatic with a nucleolus and eosinophilic cytoplasm. Cross striations within the cytoplasm can be demonstrated with a phosphotungstic acid hematoxylin stain. The cells will usually immunostain for desmin, actin (HHF-35), sarcomeric actin, troponin-T, and vimentin. Treatment is surgical resection, and according to Schiavetti and colleagues,235 it is usually combined with chemotherapy and radiation therapy. McDermott and colleagues178 pointed out that these patients were predisposed to develop cerebral metastases. The patient of Comin and colleagues43 underwent a left pneumonectomy followed by radiation. He was alive and free of disease at 9 months. In the younger age group, approximately one-third of the patients died or were living with disease, and approximately two-thirds were alive with no evidence of disease at varying periods of follow-up. Noda and coworkers196 noted that serum neuron-specific enolase was helpful in detecting metastasis and disease recurrence in a patient with alveolar rhabdomyosarcoma of the lung.
Malignant Fibrous Histiocytoma
A malignant fibrous histiocytoma is usually found in the extremities or retroperitoneum in adults. It occurs infrequently in the lung and is less common than pulmonary fibrosarcomas and leiomyosarcomas. Yousem and Hochholzer279 reviewed 22 cases that they identified from the files of the Armed Forces Institute of Pathology. McDonnell and colleagues179 reported one case of pulmonary malignant fibrous histiocytoma and reviewed 15 other cases they found in the literature. Halyard and colleagues93 reported 4 cases and reviewed 49 cases that have appeared in the English-language literature. The patients in these three reports ranged in age from 18 to 80 years, with an average age of 55 years. There was a slight preponderance of men to women (30:23). The most common clinical symptoms were cough, chest pain, weight loss, and hemoptysis. Hypoglycemia and hypertrophic pulmonary osteoarthropathy also were observed in a few patients. Hermann and associates107 reported on a 57-year-old man with spontaneous hypoglycemia associated with this tumor, with a markedly high level of insulin growth factor 2 (IGF-2), suppressed levels of insulin, as well as C-peptide associated with low levels of growth hormone and IGF-1. Following resection of the tumor, insulin and C-peptide both returned to normal and the IGF-1/IGF-2 ratio increased to the normal reference range. In most instances, the chest radiograph showed a mass lesion, usually a large solid noncavitary mass. Calcification in the mass is rarely seen. The tumors appear to be distributed randomly between both lungs. Microscopically, most of the tumors were storiform (pleomorphic), and a few were of the myxoid or inflammatory type. Tian and associates251 reported seven cases seen in 11 years in their thoracic unit in Shanyang, China. The findings were similar in all respects to those in the aforementioned reports.
The primary therapeutic approach to these tumors is complete surgical excision followed by radiation therapy or chemotherapy if either is clinically indicated. Saga and colleagues231 suggest the neoadjuvant as well as the postoperative use of chemotherapy (i.e., cisplatin with vindesine) in patients with large bulky lesions. Poor prognostic indicators are an advanced stage at the time of diagnosis, extension of the tumor into the chest wall or mediastinum, metastasis beyond the thorax, or incomplete excision. Halyard and associates93 reported that eight patients in their report had survived more than 5 years after surgical excision with or without adjuvant therapy, for a survival rate of 15%. Tian and colleagues251 reported a similar survival rate; 14% (one patient) at 48 months in seven patients after surgical resection of the tumor.
Pulmonary Chondrosarcoma
Morgan and Salama188 reported a case of pulmonary chondrosarcoma, an extremely rare tumor, and reviewed the literature. The eight patients identified with primary pulmonary chondrosarcoma ranged in age from 23 to 73 years, with an average age of 46 years. The lesions were distributed equally between the sexes. Colby and coinvestigators42 noted that fewer than 20 chondrosarcomas of the lung have been reported. The clinical symptoms in the order of frequency were cough, chest pain, and dyspnea. The tumor seemed to be more common in the left lung. Radiologically, these tumors have shown areas of calcification or ossification. These tumors were solid masses, but Parker and coworkers204 described a patient in whom the CT scan and magnetic resonance imaging (MRI) mimicked a bronchogenic cyst. Grossly, these tumors had a gray surface, were circumscribed, and appeared to have a capsule. Microscopically, the tumors contained areas of malignant cartilage with calcification or ossification. These tumors are classified histologically as (a) conventional (hyaline and/or myxoid), (b) clear cell, (c) dedifferentiated, and (d) mesenchymal. In the lung, most of these tumors have been of the conventional type. The occurrence of the other types is infrequent. The mesenchymal variant is composed of well-differentiated cartilage that may undergo calcification or even ossification, surrounded by sheets of hyperchromatic malignant small cells (blue cells) with evident staghorn vascular spaces. The mesenchymal type tends to have a more aggressive course than the other histologic types.
Nonetheless, the outlook is poor in most patients. Local recurrence and distant metastases commonly occur, as noted by Morgenroth189 and Hayashi104 and their colleagues, respectively. Huang and associates115 reported the successful surgical resection of mesenchymal chondrosarcoma of the lung and noted the immunohistochemical features of diffuse positivity against vimentin for all components and reaction to S-100 limited to the cartilaginous areas. The tumor was negative to cytokeratins, epithelial membrane antigen (EMA), leukocyte common antigen, and a panel of neuroendocrine markers. The only other case of mesenchymal chondrosarcoma of the lung in the literature that we could find was that reported by Kurotaki and colleagues.149
Pulmonary Osteosarcoma
Primary pulmonary osteosarcoma is rare. Loose and colleagues163 reported two cases and found nine other cases in the literature. For the lesion to be considered an extraosseous osteosarcoma, they adhered to the following criteria: (a) the tumor must be composed of a uniform pattern of sarcomatous tissue that excludes the possibility of a malignant mixed mesenchymal tumor, (b) osteoid or bone must be formed by the sarcoma, and (c) a primary osseous tumor must be excluded. The patients ranged in age from 35 to 83 years, with a mean of 61 years. The tumors were seen equally in men and women. The most common clinical symptom was chest pain. The sarcomas were distributed approximately equally in the right and left lungs. When possible, the tumor should be resected. The prognosis is poor. Seven patients in the collected series died of their disease, two patients died of other diseases, and three patients were alive within 2 to 14 months of follow-up. Several additional case reports of osteosarcomas have appeared in the literature in the early 1990s. Petersen206 reported on a 70-year-old man with a large lung mass. A technetium 99m–methylene diphosphonate bone scan revealed an abnormal area in the region of the left lower lung, but not in the skeleton. After lobectomy, the lesion was diagnosed as an osteosarcoma. Connolly and associates44 described a 93-year-old man whose chest radiograph showed a densely calcified lung lesion. On needle biopsy, it proved to be an osteosarcoma. The patient died at home, and the family refused an autopsy. Bhalla and colleagues16 described a 58-year-old man with a cavitary lesion that was thought to be an abscess. The CT scan showed an irregular cavity with a partially calcified thick wall. A repeat scan 3 weeks later showed increasing calcifications and a marked increase in size. The patient was treated with drainage and antibiotics but without improvement. At autopsy, the mass was a pulmonary osteosarcoma. Chapman and coworkers37 reported a case of pulmonary osteosarcoma in a 33-year-old woman. She survived for 42 months after pneumonectomy, adjuvant chemotherapy, and irradiation but developed widespread metastatic disease. Her tumor showed an overexpression of bcl-2, which is an antiapoptotic protein, and cyclin D1, which drives cells from the G1 phase of the cell cycle to the S phase. Both markers have been associated with resistance to chemotherapy. In addition, the tumor showed a higher level of genomic aberrations than do skeletal osteosarcomas.
Pulmonary Liposarcoma
A primary liposarcoma is one of the rarer sarcomas that occur in the lung. Krygier and associates147 described a patient with a pleomorphic liposarcoma whose disease ran a rapidly fatal course despite aggressive treatment. These investigators also noted 11 other cases of primary liposarcomas of the lung that had been reported in the literature. The most common type of liposarcoma was the myxomatous variety, as reported by Hochberg and Crastnopol112 and others. The most successful treatment is complete surgical resection when possible.
Synovial Sarcoma
Zeren284 and Essary68 and their associates reported a study of 25 and 12 cases, respectively, of primary pulmonary sarcomas with features of monophasic synovial sarcoma. In both studies, these tumors were seen slightly more often in women than in men. The majority of patients in Zeren and colleagues’284 study were middle-aged adults (30 to 50 years of age), but in Essary and associates’68 study the patients ranged in age from 20 to 72 years, with a median age of 31 years. The tumors varied in size, with a median of 4.2 cm. The clinical symptomatology consisted of chest pain, cough, dyspnea, and hemoptysis.
The tumors were either peripheral or central in location and appeared to be well circumscribed but were not encapsulated. The tumors had histologic and ultrastructural features of monophasic synovial sarcoma. Immunohistochemical studies revealed the tumor cells to react positively to vimentin, cytokeratins, and EMA. Tornkvist and colleagues253 discussed the unique chromosomal translocation in synovial sarcoma. The translocation is t(X;18)(p 11.2;q 11.2) resulting from the fusion of the SYT gene on chromosome 18 with the SSX gene on chromosome X. The fusion is known as the SYT-SSX fusion.
Kaplan and colleagues126 presented two similar cases. In each (one a 12-year-old girl and the other a 40-year-old woman), the researchers also described the presence of the specific chromosome translocation t(X; 18), the SYT-SSX fusion. More recently Terasaki250 and Hisaoka111 and their coworkers reported three cases of primary synovial sarcoma of the lung that were confirmed by finding SYT-SSX fusion gene transcripts. In all patients surgical removal is suggested as the treatment of choice. The prognosis of the patients with this rare tumor is undetermined but is suspected to be poor.
Kaplan and colleagues126 presented two similar cases. In each (one a 12-year-old girl and the other a 40-year-old woman), the researchers also described the presence of the specific chromosome translocation t(X; 18), the SYT-SSX fusion. More recently Terasaki250 and Hisaoka111 and their coworkers reported three cases of primary synovial sarcoma of the lung that were confirmed by finding SYT-SSX fusion gene transcripts. In all patients surgical removal is suggested as the treatment of choice. The prognosis of the patients with this rare tumor is undetermined but is suspected to be poor.
Neurogenic Sarcoma
A neurogenic sarcoma is a malignant proliferation of Schwann cells. Other synonyms for this lesion include malignant schwannoma and neurofibrosarcoma. Roviaro and colleagues227 described an example of a primary pulmonary malignant schwannoma in a 27-year-old man. The patient was symptomatic, with weight loss and chest pain. A large well-defined mass in the right lower lung field was identified on radiographic studies of the chest. Histologically the tumor consisted of immature fusiform cells in sheets and cords. Mitotic figures were common. Some of the cells tended to form fascicles with a reticular pattern resembling that of a neurofibroma. The final diagnosis was that of a malignant schwannoma. Recurrence of the tumor occurred shortly after resection, and the patient died of the malignant process 4 months after the surgical resection. A second patient was reported by Rowlands and associates.228 A primary malignant melanotic schwannoma of the right upper lobe bronchus was identified in a 27-year-old man. A sleeve lobectomy was performed, and on histologic examination the morphology of the tumor was characteristic of a melanotic schwannoma. Areas of both Antoni A and B pattern were present, and varying numbers of melanosomes at different levels of maturation were readily demonstrated. Mitotic figures were scanty. The patient developed a cerebral metastasis and was terminal 14 months after the initial diagnosis.
McCluggage and Bharucha175 subsequently reported two additional cases and reviewed the literature. Their patients were 34 and 45 years of age; one was a man and the other a woman. Both presented with dyspnea and chest pain. On gross examination, one tumor was 8 cm and the other 10 cm in diameter. One was a white, circumscribed mass that had a whirled appearance on cut section. The other was also white but contained necrotic areas. On microscopic examination, the smaller tumor had a benign appearance with a fibrous capsule and spindle cells with irregular wavy nuclei. The larger tumor was highly cellular with necrosis. It had pleomorphic cells and multinucleated giant cells with easily identified mitotic figures. Immunostaining revealed that both tumors were focally positive for S-100 protein and diffusely positive for vimentin. They were negative for desmin, carcinoembryonic antigen, and CAM 5.2, a keratin. The tumor was resected in both patients, but each patient subsequently developed metastatic disease.
Primary Pulmonary Ganglioneuroblastoma
Primary ganglioneuroblastoma of the lung is rare. Only three cases have been reported in the English-language literature. Cooney45 reported the first case, which occurred in a 47-year-old man. The patient presented with cough and was found to have a 5-cm mass in the right lower lobe on radiographs of the chest. The tumor was removed by a bilobectomy of the middle and right lower lobes. On examination of the specimen, the tumor abutted the segmental bronchi, but no intrabronchial tumor was noted. Microscopically the tumor had a fibrous capsule that enclosed a rim of mature ganglioneuromatous tissue containing mature and immature ganglion cells that surrounded a central core composed of primitive neuroblastoma. Vascular invasion of the tumor was evident. Cooney suggested that the tumor arose within a sympathetic component of the posterior pulmonary plexus. The patient was living and well 2½ years after the surgical resection.
The second and third cases of primary pulmonary ganglio- neuroblastoma were recorded by Hochholzer and colleagues.113 Both patients were adult women, and in each the tumor extended from an adjacent bronchus. The first patient was 38 years old and had the signs and symptoms of an advanced multiple endocrine neoplasia (MEN) type 1 syndrome. Blood chemistries supported the diagnosis. There were also radiographic findings of a 3-cm infrahilar mass and two small peripheral nodules in the right lung. The patient died within several days of admission, and the autopsy revealed the perihilar mass to be a typical ganglioneuroblastoma with direct invasion of two adjacent hilar lymph nodes but no intrabronchial extension. One of the peripheral masses was a carcinoid tumorlet and the other was a metastatic islet cell tumor of the pancreas. An islet cell tumor of the tail of the pancreas, a tumor of a parathyroid gland, and a cystic tumor of the pituitary gland were also present. The thyroid gland was normal. The second patient was 20 years old and essentially asymptomatic, but radiographs of the chest revealed a mass in the left upper lobe. A left upper lobectomy was performed and the mass, 5 by 5 cm in size, histologically was a typical ganglioneuroblastoma abutting and invading the bronchial lumen. No lymph node involvement was seen. Immunohistochemical studies revealed focal staining for neurofilament protein and S-100 protein and diffuse staining for neuron-specific enolase. The staining for chromogranin, keratin, and glial fibrillary acid protein was negative. The patient was alive without metastatic or locally recurrent disease 1 year after the surgical resection.
Although the eventual prognosis has yet to be determined, the primary pulmonary neuroblastomas to date appear to have a low malignant potential. Surgical resection is the treatment of choice.
Primary Malignant “Triton” Tumor of the Lung
A malignant neurogenic tumor with rhabdomyoblastic differentiation is known as a triton tumor (ectomesenchymoma). These are more common in the soft tissues, and their occurrence in the lung is rare. In the thorax these tumors are more commonly located in the mediastinum. Moran and colleagues187 have described two such tumors that originated in the lung. One patient was a 3-year-old child and the other a 53-year-old man. Both patients had a large intrapulmonary mass, and each presented with marked dyspnea. Both lesions were removed by pneumonectomy. The tumors were characterized by atypical spindle-cell proliferation in an abundant myxoid stroma. Areas of focal rhabdomyoblastic differentiation were present and were characterized by large cells with occasional cytoplasmic cross striations. Immunohistochemically, focal reaction to S-100 protein
was present in the atypical spindle cells and a strong reaction to desmin and myoglobin was present in the rhabdomyoblastic areas. The course of the tumor was rapidly fatal in the child, and there is no follow-up information on the man.
was present in the atypical spindle cells and a strong reaction to desmin and myoglobin was present in the rhabdomyoblastic areas. The course of the tumor was rapidly fatal in the child, and there is no follow-up information on the man.
Malignant Mesenchymoma
Malignant mesenchymoma is a sarcoma composed of two or more cellular elements, excluding fibrous tissue. Domizio and colleagues61 reported a case arising from a cyst in a 4-year-old boy. The boy had a history of pulmonary cyst in the right lower lobe diagnosed when he was 6 months old by chest radiograph; he had a clinical history of anorexia, recurrent dry cough, and night sweats. He was anemic with an elevated erythrocyte sedimentation rate. At the time of admission, the chest radiograph showed an opaque right hemithorax with an air-fluid level in the right midlung field and the mediastinum shifted to the left. He underwent a right lower lobectomy. On gross examination, the lobe was replaced by a necrotic tumor. The viable outer rim was composed of yellowish-gray gelatinous nodules with an area of central necrosis. Microscopically, the tumor was composed of large anaplastic cells with numerous multinucleate giant cells. There were areas of rhabdomyosarcoma and chondrosarcoma. A cystic lesion was also present with multiple cystic spaces lined by epithelial cells. The patient was treated with chemotherapy and radiation therapy and was free of disease at short-term follow-up.
Sarcomas of Large Vessel Origin
A pulmonary trunk sarcoma is a primary lesion arising within the pulmonary artery or, as Mandelstramm168 described, from the pulmonary valve of the heart. In reviews by Wackers and colleagues,263 Bleisch and Kraus,17 Baker and Goodwin,10 and Goldblum and Rice86 as well as by Emmert-Buck63 and Nonomura199 and their associates, undifferentiated sarcoma, leiomyosarcoma, and fibrosarcoma make up the majority of the cell types of these intravascular tumors but also include pleomorphic rhabdomyosarcoma and epithelioid angiosarcoma. Burke and Virmani,25 after doing immunohistochemistry, concluded that most pulmonary artery sarcomas were derived from intimal cells with myofibroblastic differentiation. Ko138 and Leone155 and their coworkers both described leiomyosarcomas in the pulmonary vein. In the past several years, additional case reports of large vessel sarcomas have been published (see reading references at the end of this chapter).
The large vessel sarcomas may spread distally within the vascular tree or extend outside the vessel to invade the lung tissue. The patients may be of any age. In reported cases, the age range was 21 to 81 years, with an average age of 50 years. There is a slight predominance of these large vessel sarcomas in women. The patients present with chest pain and dyspnea, and one third may also have cough, hemoptysis, and palpitations. A systolic heart murmur may be present. Pulmonary hypertension with proximal dilatation of the vessels is a constant feature. A late manifestation is right-sided heart decompensation. Moffat and colleagues184 reported the radiographic features, as did Britton.24 The lesion manifests most often as a lobulated perihilar mass. Angiography may reveal multiple defects within the pulmonary artery. CT and MRI may help to determine the extent of the disease. Mader and colleagues (1997) believe that MRI is the imaging modality of choice for these lesions because it is noninvasive and gives an excellent definition of the heart, pericardium, mediastinum, and lungs. MRI can also delineate both the extent and location of the lesion. Cox and colleagues48 agree with this assessment and believe the imaging findings are quite specific. Parish and associates,202 in addition to discussing the MRI and CT features of nine cases, suggest that transesophageal echocardiography might also be useful in evaluating pulmonary trunk sarcomas. Simpson and Mendelson243 agree that the new imaging modalities, including helical CT and MRI, are useful in making premortem diagnosis. They describe three cases. Treatment is resection. Head105 and Redmond219 and their coworkers, among others, reported cases in which these tumors were successfully resected. Kruger and associates145 reported prolonging survival with resection followed by adjuvant therapy; however, the prognosis for long-term survival is poor. Genoni and colleagues84 describe four patients who underwent resection for pulmonary trunk sarcomas. One had a thromboendarterectomy of the pulmonary trunk with adjuvant radiation therapy and remains disease-free after 3 years. One tumor was resected, but the patient developed metastases. After chemotherapy, the metastases disappeared and the patient was alive and well 1 year later. The other two patients died within 2 months of surgery. One died as the result of a tumor mass in the inferior vena cava and the other died of cerebral metastases. Mayer and coworkers174 reported the results of resection in seven patients with pulmonary artery sarcomas. None of the patients died in the perioperative period. Four patients were dead after 19 months due to metastatic disease or recurrent tumor; two patients were alive after 21 and 35 months despite the presence of metastatic pulmonary disease. However, one patient was alive at 62 months with no evidence of recurrent disease.
Sarcomas of Small Vessel Origin
Angiosarcoma, epithelioid hemangioendothelioma, and hemangiopericytoma are malignant vascular tumors that occasionally may occur in the lung but are rare in this location. Kaposi’s sarcoma, a vascular neoplasm, is not discussed here because it has not been described as having a primary pulmonary origin. Last, according to Enzinger and Weiss,83 the term hemangio- endothelioma should be used only to designate a group of vascular tumors that cannot be accurately classified histologically as to their ultimate biological behavior.
Angiosarcoma
An angiosarcoma is a malignant neoplasm of endothelial cells. It has also been called a malignant hemangioma or malignant hemangioendothelioma. When the neoplastic endothelial cells have an epithelial appearance, the sarcomas are referred to as epithelioid angiosarcomas. Patel and Ryu205 reviewed the files of the Mayo Clinic from 1950 to 1990 and could not identify any primary pulmonary angiosarcomas. They identified 15 patients with metastatic angiosarcoma and discussed the single case reports of so-called primary angiosarcoma. Sheppard and colleagues237 described an epithelioid angiosarcoma of the lung in a 65-year-old man who presented with pulmonary hemorrhage. At autopsy, the lungs were hemorrhagic, with multiple nodules of tumor. An angiosarcoma can be associated with a hemothorax, hypertrophic pulmonary osteoarthropathy, or both. Atasoy
and coworkers7 reported a primary pulmonary angiosarcoma. In their report, they pointed out that MRI showed a heterogeneous pattern consisting of hyperintense areas scattered throughout a background of intermediate signal intensity. These findings gave the lesion a cauliflower-like appearance, especially on T2-weighted images. The prognosis for patients with angiosarcoma is poor.
and coworkers7 reported a primary pulmonary angiosarcoma. In their report, they pointed out that MRI showed a heterogeneous pattern consisting of hyperintense areas scattered throughout a background of intermediate signal intensity. These findings gave the lesion a cauliflower-like appearance, especially on T2-weighted images. The prognosis for patients with angiosarcoma is poor.
Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma is a low-grade sclerosing angiosarcoma that occurs in the lung as well as the liver, bone, soft tissue, and other sites. This vascular lesion was first described by Dail and Liebow75 in 1975. Dail and colleagues55 reviewed an additional 19 cases. They initially called this tumor an intravascular bronchioloalveolar tumor (IVBAT), but they now prefer the term sclerosing endothelial tumor. Weiss and Enzinger268 described 41 cases of an identical tumor occurring in soft tissue and proposed the name epithelioid hemangioendothelioma, which has become widely accepted. Weiss and colleagues269 published a combined review of lesions in soft tissue, lung, liver, and bone. Since then, individual case reports have appeared in the literature. Wenisch and Lulay270 reported that, in the lung, these tumors occur in patients who are 4 to 70 years of age, with one-third of the patients less than 30 years of age. The tumor occurs four times more frequently in women than in men. Most of the patients are asymptomatic or complain of a nonproductive cough. Ross and associates223 note that the chest radiographs and CT scans reveal many small (<1 cm in diameter) nodular densities in both lung fields. According to Moran and Suster,185 the tumors are characterized by proliferation of round-to-oval epithelial endothelioid cells. These cells contain abundant cytoplasm with oval nuclei embedded in a hyaline matrix. The tumor has a tendency to fill the alveoli in a polypoid fashion. The cells are immunohistochemically positive for endothelial markers (factor VIII–related antigen, Ulex europucus lectin, CD31, and CD34). Weibel-Palade bodies may be seen infrequently on electron microscopic examination. The average survival after diagnosis is 4.6 years. Lymph node metastasis in uncommon. Cronin and Arenberg49 found only a 9% incidence to be present in their review of the literature. On the other hand, distant metastasis is more common. Bagan and colleagues9 reported an incidence of 39% in 31 cases in their report. The liver was involved in six, bone in three, the brain in two and the bowel in one patient. Kitaichi and coworkers137 reviewed 21 patients with epithelioid hemangioendothelioma. They found three subsets of patients who had a poor prognosis. These patients had either pleural effusions, tumors with a spindle-cell component, or fibrinofibrous pleuritis with the presence of extrapleural tumor cells. Bagan and coworkers9 reviewed the findings of 75 patients in the English and French literature and added five cases of their own. The data were essentially the same as just presented. They did, however, separate the patients into two groups: (a) asymptomatic with only a pulmonary nodule or nodules and (b) symptomatic patients due to vascular endothelial cell proliferation resulting in alveolar hemorrhage, hemoptysis, hemorrhagic pleural effusion or the presence of anemia. Patients in the first group had a satisfactory prognosis with a median survival of 180 months; patients in the second group had a much poorer survival. Of all of the poor prognostic features, the presence of bilateral hemorrhagic effusion was the worst. This was amply illustrated by the case reported by Rossi and associates225 in which a 14-year-old child died within 3 months of the diagnosis.
Treatment should be governed by the extent of the disease in the lungs and the presence or absence of distant metastasis, especially if the liver is involved. Lerut and coworkers156 have reported satisfactory outcomes following liver transplantation in selected patients. Bagan and colleagues9 suggest the possibility of lung transplantation in highly selected patients with extensive bilateral pulmonary disease.
Bagan and coworkers9 briefly discussed the use of various drugs in the management of these tumors. Interferon α2a, azathioprine, steroids, carboplatin, and etoposide have been used with variable and unpredictable results. Death from pulmonary insufficiency is the usual course of this disease, although distant metastatic disease may likewise be a relatively common cause.
Hemangiopericytomas
Hemangiopericytomas are unusual sarcomas derived from the ubiquitous capillary pericytic cell and are commonly located in the soft tissues of the thigh and retroperitoneum. Yousem and Hochholzer280 found that pulmonary hemangiopericytomas occurred with equal frequency in men and women; the average age of patients was 46.1 years. Approximately one-third of the patients were asymptomatic. Those with symptoms complained of chest pain, hemoptysis, dyspnea, and cough. One patient had pulmonary osteoarthropathy. Radiographs of the chest usually show a lobulated, well-circumscribed, homogeneous soft tissue density, but other findings may also be present (Fig. 125-3). Rusch and colleagues230 found that MRI was critical in the preoperative evaluation of these patients because of its ability to delineate the anatomic extent of the lesion. Treatment is surgical excision. Prognosis is variable. Indicators of a poor prognosis are chest symptoms, tumor >8 cm in diameter, pleural and bronchial wall invasion, tumor giant cells, more than three mitoses per 10 high-power fields (HPFs), and tumor necrosis. Shin and Ho241 noted that 33% of patients with tumors ≥5 cm had metastasis and 66% of those with tumors ≥10 cm had metastasis. Davis and coworkers56 demonstrated that most recurrences took place within 2 years of diagnosis. Enzinger and Smith66 and Feldman and Seaman74 reported that chemotherapy and radiation therapy do not consistently help the patient. In separate case reports, Wu,277 Shimizu,238 Kiefer,132 Van Damme,262 Rusch,230 Hansen,97 and Brega Massone23 and their colleagues described their experience with pulmonary hemangiopericytomas. The patients described by Van Damme and coworkers262 died within several months of their operation. In contrast, the patient of Rusch and associates230 was disease-free 28 months after surgical excision. Wu and colleagues277 described a patient who had an associated coagulopathy that recurred when the tumor recurred.
Carcinosarcoma
The terminology of carcinosarcoma has been changing as pathologists examine these neoplasms more closely. A carcinosarcoma is defined as a malignant neoplasm composed of both malignant epithelial and mesenchymal elements. The mesenchymal component should show differentiation into specific
heterologous mesenchymal tissues, such as bone, cartilage, or striated muscle, but not fibroblasts. The purpose of this definition was to separate carcinosarcoma from spindle cell carcinoma. Nappi and associates194 reviewed 21 cases of carcinosarcomas and spindle cell carcinomas. They believed that these two neoplasms were merely part of a spectrum and suggested adopting the names biphasic sarcomatoid carcinoma for carcinosarcoma and monophasic sarcomatoid carcinoma for spindle-cell carcinoma. They noted that both of these tumors tended to behave in an aggressive fashion, with 20 of their patients dying within 2 years and only 1 patient alive with no evidence of disease at 21 months. Ishida and colleagues118 described five cases of carcinosarcoma and three cases of spindle cell carcinoma. Kimino and coworkers134 described a case in which the sarcomatous component was positively stained by the EMA, suggesting a sarcomatous transformation of the carcinomatous component of the tumor. Grahmann and coworkers90 reported three cases of carcinosarcoma and reviewed the literature, as did Berho14 and Wick271 and their colleagues. Wick and colleagues271 went on to place both these tumors under the designation sarcomatoid carcinoma.
heterologous mesenchymal tissues, such as bone, cartilage, or striated muscle, but not fibroblasts. The purpose of this definition was to separate carcinosarcoma from spindle cell carcinoma. Nappi and associates194 reviewed 21 cases of carcinosarcomas and spindle cell carcinomas. They believed that these two neoplasms were merely part of a spectrum and suggested adopting the names biphasic sarcomatoid carcinoma for carcinosarcoma and monophasic sarcomatoid carcinoma for spindle-cell carcinoma. They noted that both of these tumors tended to behave in an aggressive fashion, with 20 of their patients dying within 2 years and only 1 patient alive with no evidence of disease at 21 months. Ishida and colleagues118 described five cases of carcinosarcoma and three cases of spindle cell carcinoma. Kimino and coworkers134 described a case in which the sarcomatous component was positively stained by the EMA, suggesting a sarcomatous transformation of the carcinomatous component of the tumor. Grahmann and coworkers90 reported three cases of carcinosarcoma and reviewed the literature, as did Berho14 and Wick271 and their colleagues. Wick and colleagues271 went on to place both these tumors under the designation sarcomatoid carcinoma.
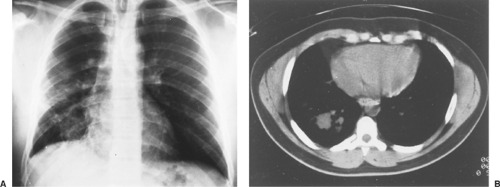 Figure 125-3. A: Posterior radiograph of the chest reveals a 5-cm mass in the right lower lobe. Suggestion of additional masses is evident on the levels of the second and third anterior interspaces. B: CT scan reveals a mass in the right lower lobe with multiple satellite lesions. Histologic examination of the resected specimen revealed a poorly differentiated hemangiopericytoma. |
Carcinosarcomas (sarcomatoid carcinomas) frequently have a slow rate of growth. Much of the growth can be endobronchial with little propensity to infiltrate the bronchial wall, but extensive invasion into the surrounding lung does occur. Metastases to the regional lymph nodes and to distant organs, especially to the brain, are common. The more common symptoms are cough and hemoptysis. Chest pain, fever, and malaise may occur. Meade and coworkers181 described a patient with associated pulmonary osteoarthropathy. Patients with a peripheral tumor may be asymptomatic. Bronchial biopsy may be followed by excessive hemorrhage but is nonetheless indicated for the preoperative evaluation of an endobronchial lesion. Surgical resection, when possible, is the indicated treatment. In most series, however, most of the patients die within the first year of resection. Approximately 16% to 23% of patients may survive 5 years or longer. However, in the series reported by Miller and Allen,182 a 5-year survival rate of only 6% was recorded. Koss and coworkers141 studied 66 cases of carcinosarcomas. The patients had a mean age of 65 years with a ratio of men to women of 7.25:1. The tumors usually presented in the upper lobes and many measured >7 cm in its greatest dimension. The majority (62%) were endobronchial or central, with 38% being peripheral in location (Fig. 125-4). The 5-year survival rate was 21.3%. Only the tumor size (>6 cm) appeared to be adversely related to survival.
Recently, several single case reports by Shah and Sabanathan236 as well as those by Kim133 and Saha and coworkers232 have been published, but no new essential information was added to our understanding of these interesting tumors. Rossi and associates224 recorded three cases in their report. The sarcomatous component was cartilage in two cases and bone in the third; the carcinomatous foci were squamous cell carcinomas in all three.
Uncommon Carcinomas of the Lung
Carcinomas with Pleomorphic, Sarcomatoid, or Sarcomatous Elements
The World Health Organization (WHO)/International Association for the Study of Lung Cancer276 categorized a group of tumors as carcinomas with pleomorphic, sarcomatoid, or sarcomatous elements. The included tumors are listed in Table 125-3. In Rossi and colleagues’224 series of 75 patients in this category, 58 had a pleomorphic carcinoma; 7 had only a mixture of spindle and giant cells in the tumor; 10 had a pure spindle-cell cancer; 3 had only giant cells present; 3 of the remaining 4 patients had a carcinosarcoma; and 1 had a pulmonary blastoma.
Pleomorphic Carcinoma of the Lung
The WHO classification276 of tumors identified a pleomorphic carcinoma as a specific type of lung cancer with pleomorphic, sarcomatoid, or sarcomatous elements (spindle- or giant-cell
carcinomas or both) combined with squamous cell carcinoma, an adenocarcinoma, or a large-cell carcinoma. To qualify as a pleomorphic tumor, the spindle-cell carcinoma or the giant cell carcinoma component should constitute at least 10% of the neoplasm’s mass. Pure spindle- and giant-cell carcinomas are thought to be rare. Areas of necrosis are common, according to Chang and coworkers.35 The tumors express focal staining for cytokines and a diffused stain form vimentin. Rossi and colleagues224 utilized the thyroid transcription factor-1 (TTF-1) and CK 7 to verify the pulmonary origin of these tumors. Fifty-five percent of 20 cases were positive for TTF-1 and 70% were positive for CK staining; surfactant protein A staining was negative in all.
carcinomas or both) combined with squamous cell carcinoma, an adenocarcinoma, or a large-cell carcinoma. To qualify as a pleomorphic tumor, the spindle-cell carcinoma or the giant cell carcinoma component should constitute at least 10% of the neoplasm’s mass. Pure spindle- and giant-cell carcinomas are thought to be rare. Areas of necrosis are common, according to Chang and coworkers.35 The tumors express focal staining for cytokines and a diffused stain form vimentin. Rossi and colleagues224 utilized the thyroid transcription factor-1 (TTF-1) and CK 7 to verify the pulmonary origin of these tumors. Fifty-five percent of 20 cases were positive for TTF-1 and 70% were positive for CK staining; surfactant protein A staining was negative in all.
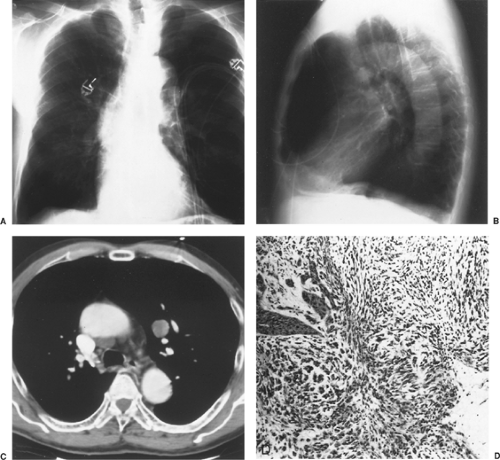 Figure 125-4. Postanterior (A) and left lateral (B) chest radiographs of a 75-year-old man with suspected myocardial infarction. An asymptomatic 3.2-cm mass is identified just below the aortic arch. CT scan (C) demonstrates a solitary parenchymal lung mass. Photomicrograph (D) after its removal by a left upper lobectomy shows that the lesion is a carcinosarcoma of the lung. |
Table 125-3 Carcinomas With Pleomorphic, Sarcomatoid, or Sarcomatous Elements | |||||
|---|---|---|---|---|---|
|
Table 125-4 Histologic Composition of Pleomorphic Carcinomas (PCs) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ro,221 Fishback,77 Chang,35 Rossi,224 and Raveglia217 and their associates have described 12, 78, 16, 58, and 20 cases of pleomorphic carcinomas, respectively (Table 125-4). As a rule, the non-small-cell carcinomatous elements are sparse in relationship to the tumors’ volume. The non-small-cell carcinomas are as noted previously. The incidence of each may vary in any given report (see Table 125-4). The occurrence of a small-cell cancer is very rare; Fishback and colleagues77 have recorded only one, and in that patient there was accompanying adenocarcinoma in the tumor. The presence of two of the three non-small-cell cancers in one tumor is not uncommon.
Ro and associates221 describe the sarcomatous portions of these tumors as resembling a malignant fibrous histiocytoma, a fibrosarcoma, or an unclassified sarcoma. Raveglia and coworkers217 noted a mixture only of giant cells and sarcomatous cells in 14 of the 20 tumors they examined. Other investigators including Fishback77 and Rossi224 and their associates have reported similar finding but with less frequency. The first aforementioned group reported 17 such tumors in 78 patients and the second group recorded 7 such tumors in 58 patients, respectively.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


