TUMORS AND TRAUMA OF THE HEART
Eric H. Awtry ![]() Wilson S. Colucci
Wilson S. Colucci
TUMORS OF THE HEART
PRIMARY TUMORS
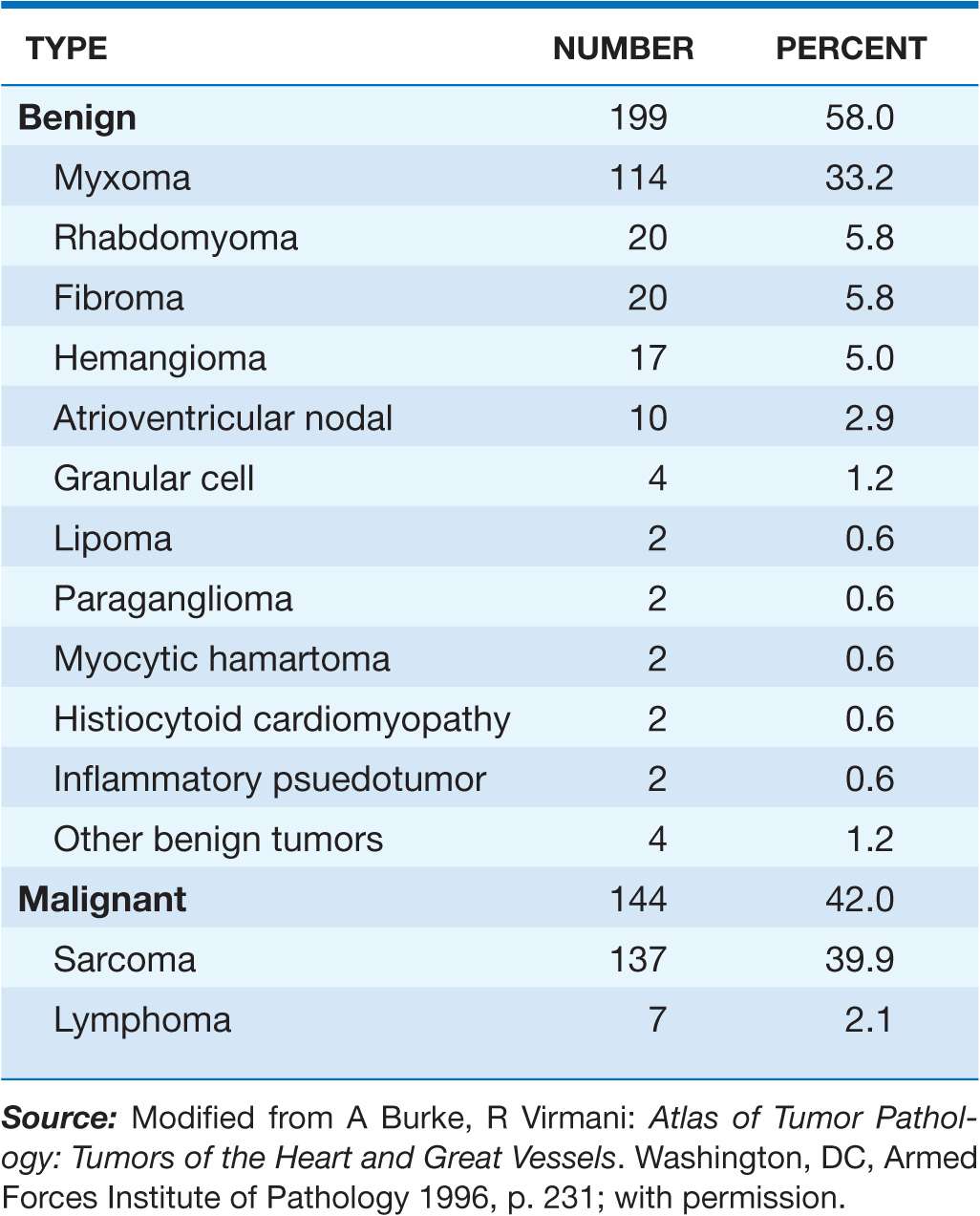
Primary tumors of the heart are rare. Approximately three-quarters are histologically benign, and the majority of these tumors are myxomas. Malignant tumors, almost all of which are sarcomas, account for 25% of primary cardiac tumors (Table 23-1). All cardiac tumors, regardless of pathologic type, have the potential to cause life-threatening complications. Many tumors are now surgically curable; thus, early diagnosis is imperative.
TABLE 23-1
RELATIVE INCIDENCE OF PRIMARY TUMORS OF THE HEART
Clinical presentation
Cardiac tumors may present with a wide array of cardiac and noncardiac manifestations. These manifestations depend in large part on the location and size of the tumor and are often nonspecific features of more common forms of heart disease, such as chest pain, syncope, heart failure, murmurs, arrhythmias, conduction disturbances, and pericardial effusion with or without tamponade. Additionally, embolic phenomena and constitutional symptoms may occur.
Myxoma
Myxomas are the most common type of primary cardiac tumor in all age groups, accounting for one-third to one-half of all cases at postmortem and about three-quarters of the tumors treated surgically. They occur at all ages, most commonly in the third through sixth decades, with a female predilection. Approximately 90% of myxomas are sporadic; the remainder are familial with autosomal dominant transmission. The familial variety often occurs as part of a syndrome complex (Carney complex) that includes (1) myxomas (cardiac, skin, and/or breast), (2) lentigines and/or pigmented nevi, and (3) endocrine overactivity (primary nodular adrenal cortical disease with or without Cushing’s syndrome, testicular tumors, and/or pituitary adenomas with gigantism or acromegaly). Certain constellations of findings have been referred to as the NAME syndrome (nevi, atrial myxoma, myxoid neurofibroma, and ephelides) or the LAMB syndrome (lentigines, atrial myxoma, and blue nevi), although these syndromes probably represent subsets of the Carney complex. The genetic basis of this complex has not been elucidated completely; however, patients frequently have inactivating mutations in the tumor-suppressor gene PRKAR1A, which encodes the protein kinase A type I-α regulatory subunit.
Pathologically, myxomas are gelatinous structures that consist of myxoma cells embedded in a stroma rich in glycosaminoglycans. Most are solitary, are located in the atria (particularly the left atrium, where they usually arise from the interatrial septum in the vicinity of the fossa ovalis), and are often pedunculated on a fibrovascular stalk. In contrast to sporadic tumors, familial or syndromic tumors tend to occur in younger individuals, are often multiple, may be ventricular in location, and are more likely to recur after initial resection.
Myxomas commonly present with obstructive signs and symptoms. The most common clinical presentation mimics that of mitral valve disease: either stenosis owing to tumor prolapse into the mitral orifice or regurgitation resulting from tumor-induced valvular trauma. Ventricular myxomas may cause outflow obstruction similar to that caused by subaortic or subpulmonic stenosis. The symptoms and signs of myxoma may be sudden in onset or positional in nature, owing to the effects of gravity on tumor position. A characteristic low-pitched sound, a “tumor plop,” may be appreciated on auscultation during early or mid-diastole and is thought to result from the impact of the tumor against the mitral valve or ventricular wall. Myxomas also may present with peripheral or pulmonary emboli or with constitutional signs and symptoms, including fever, weight loss, cachexia, malaise, arthralgias, rash, digital clubbing, Raynaud’s phenomenon, hypergammaglobulinemia, anemia, polycythemia, leukocytosis, elevated erythrocyte sedimentation rate, thrombocytopenia, and thrombocytosis. These factors account for the frequent misdiagnosis of patients with myxomas as having endocarditis, collagen vascular disease, or a paraneoplastic syndrome.
Two-dimensional transthoracic or omniplane transesophageal echocardiography is useful in the diagnosis of cardiac myxoma and allows assessment of tumor size and determination of the site of tumor attachment, both of which are important considerations in the planning of surgical excision (Fig. 23-1). CT and MRI may provide important information regarding size, shape, composition, and surface characteristics of the tumor (Fig. 23-2).
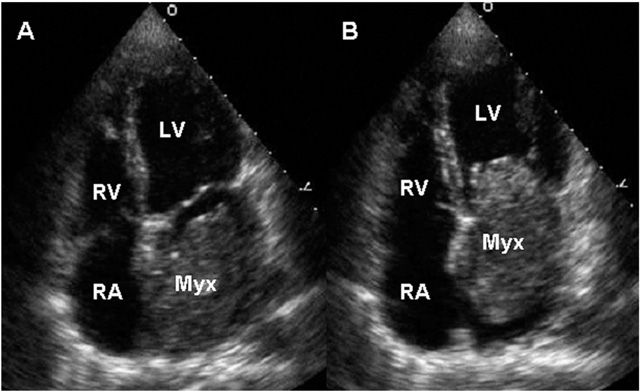
FIGURE 23-1
Transthoracic echocardiogram demonstrating a large atrial myxoma. The myxoma (Myx) fills the entire left atrium in systole (panel A) and prolapses across the mitral valve and into the left ventricle (LV) during diastole (panel B). RA, right atrium; RV, right ventricle. (Courtesy of Dr. Michael Tsang; with permission.)
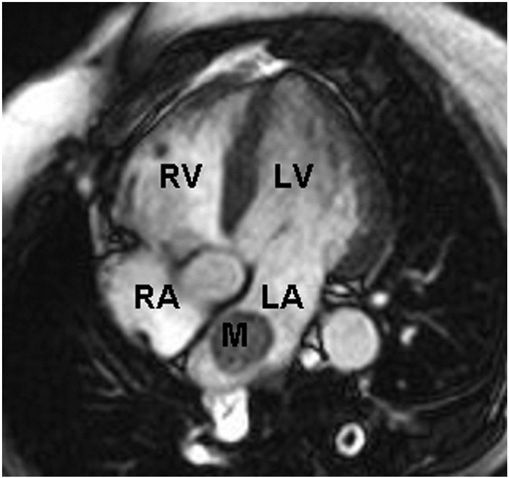
FIGURE 23-2
Cardiac MRI demonstrating a rounded mass (M) within the left atrium (LA). Pathologic evaluation at the time of surgery revealed it to be an atrial myxoma. LV, left ventricle; RA, right atrium; RV, right ventricle.
Although cardiac catheterization and angiography were previously performed routinely before tumor resection, they no longer are considered mandatory when adequate noninvasive information is available and other cardiac disorders (e.g., coronary artery disease) are not considered likely. Additionally, catheterization of the chamber from which the tumor arises carries the risk of tumor embolization. Because myxomas may be familial, echocardiographic screening of first-degree relatives is appropriate, particularly if the patient is young and has multiple tumors or evidence of myxoma syndrome.
TREATMENT Myxoma
Surgical excision utilizing cardiopulmonary bypass is indicated regardless of tumor size and is generally curative. Myxomas recur in 12–22% of familial cases but in only 1–2% of sporadic cases. Tumor recurrence most likely is due to multifocal lesions in the former and inadequate resection in the latter.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


