Chapter 30 Tumor Biology and Tumor Markers
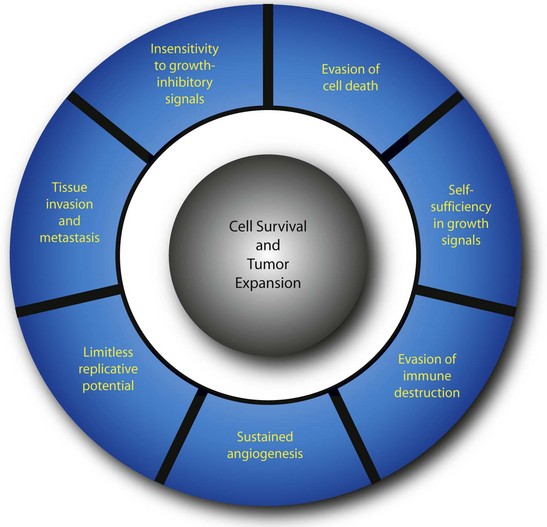
Neoplasia (literally meaning “new growth”) is the uncontrolled proliferation of transformed cells. The term tumor, which was originally used to describe the swelling caused by inflammation, is now used interchangeably with neoplasm. Transformation is the multistep process whereby normal cells acquire malignant characteristics. Each step reflects a genetic alteration that confers a growth advantage over normal cells. There are a number of essential alterations in cell physiology that collectively enable malignant growth1,2; self-sufficiency in growth signals, evasion of programmed cell death (apoptosis), evasion of immune detection and destruction, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis. These characteristics are shared by most, if not all, human tumors.
Epidemiology
Approximately 1.5 million new cases of cancer were expected to be diagnosed in 2010, apart from the more than 1 million new cases of basal and squamous cell cancers (Fig. 30-1). In men, the most common cancers are prostate, lung, colorectal, and urinary bladder cancers (Table 30-1). In women, the most common cancers are breast, lung, colorectal, and uterine (cervical and endometrial).
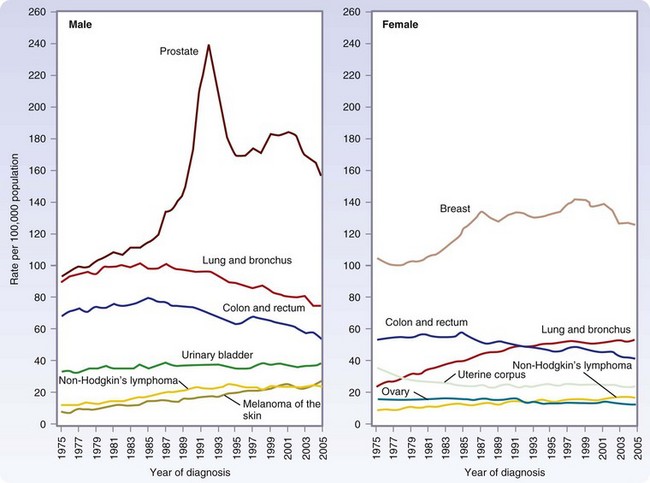
(From Jemal A, Siegel R, Ward E, et al: Cancer statistics, 2009. CA Cancer J Clin 59:225–249, 2009.)
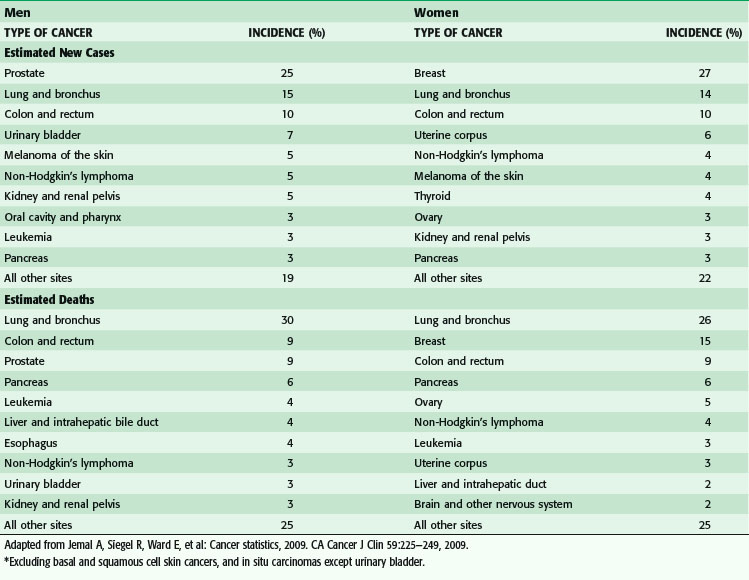
Cancer is the second most common cause of death in the United States, accounting for one of every four deaths (Fig. 30-2). In 2010, more than a half-million Americans will die of cancer.
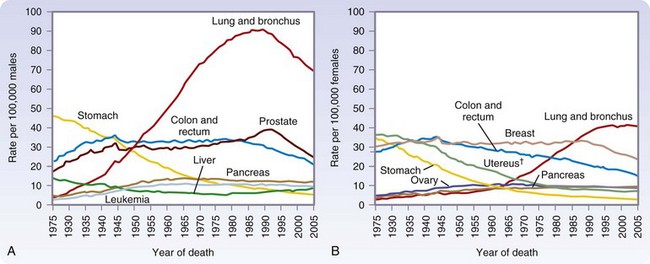
(From Jemal A, Siegel R, Ward E, et al: Cancer statistics, 2009. CA Cancer J Clin 59:225–249, 2009.)
Global Burden of Cancer
Worldwide, cancer is responsible for one in eight deaths. The distribution and types of cancer that occur continue to change, being affected primarily by the following: (1) the growth and aging of populations; (2) the increasing encroachment of modifiable risk factors (e.g., cigarette smoking, Western diet, physical inactivity) in developing countries; and (3) the relatively slower decrease in infection-related cancers.3 By 2020, 70% of all cancer-related deaths will be in developing countries, in which survival rates (20% to 30%) are barely half those of developed countries.4 Indeed, 80% to 90% of people diagnosed with cancer in developing countries present with late-stage, terminal cancer. It can therefore be seen that the vast majority of cancer deaths will occur in countries least equipped to handle the burden.
Aging and Cancer
Cancer disproportionately affects people 65 years and older. In the United States, this age group comprises 56% of all newly diagnosed cancer patients and 71% of all cancer deaths.5 The median age of death for cancers common to both men and women (including lung, colorectal, pancreas, stomach, and urinary bladder) cancers, ranges from 71 to 77 years. The number of people in this age group will double to 70 million (or one in five people) over the next 25 years, driven by the Baby Boom cohort born between 1946 and 1964. This is a recognized trend throughout the developed world.
Cancer treatment in older adults is less well-studied and it has been shown that the older population is underrepresented in clinical trials.6–8 There have been a number of reports of the underuse of adjuvant therapy, chemotherapy and radiotherapy, in the aging population. O’Connell and colleagues9 have studied the Surveillance, Epidemiology, and End Results (SEER) database (1988–1997), and found that although older patients with colorectal or breast cancer had excellent rates of receiving cancer-directed surgery, rates were variable for many other cancers, including lung, esophagus, stomach, liver, and pancreas cancers. Surgical intervention not being recommended was the most common reason. Clearly, the surgeon must more carefully weigh an individual’s operative risk in the context of the difficulty, length and morbidity of the procedure, with greater consideration for quality of life and functional status, beyond just postoperative mortality and mortality and long-term survival.
Obesity and Cancer
Prevalence of overweight (body mass index [BMI], 25 to 30) and obesity (BMI ≥ 30) in most developed countries, and in urban areas of many less developed countries, has been increasing markedly over the past 25 years. In the United States, approximately one third of the population is now classified as obese. Although obesity has long been recognized as an important cause of diabetes and cardiovascular disease, the relationship between obesity and cancer has received less attention. Epidemiologic studies indicate that adiposity contributes to the increased incidence and/or death from cancers of the colon, breast (in postmenopausal women), endometrium, kidney (renal cell), esophagus (adenocarcinoma), gastric cardia, pancreas, gallbladder, and liver (hepatocellular carcinoma). It has been estimated that 15% to 20% of all cancer deaths in the United States can be attributed to overweight and obesity.10
Tumor Biology
Much has been learned about the multistep process of tumorigenesis. A well-documented example of tumor development is presented in Table 30-2. The transformation of melanocytes into malignant melanoma can be divided histopathologically and clinically into five major identifiable steps. Successive genetic changes each confer a growth advantage, leading to the progressive conversion of normal cells into cancer cells. This process is associated with a number of distinct changes in cell physiology (Fig. 30-3),1,2 each of which is discussed in detail here.
Table 30-2 Stepwise Progression from Melanocyte to Metastatic Melanoma
| STEP* | CHARACTERISTICS |
|---|---|
| 1 | Common melanocytic nevus |
| 2 | Dysplastic nevus |
| 3 | Radial growth phase of melanoma |
| 4 | Vertical growth phase of melanoma |
| 5 | Metastatic melanoma |
* Common acquired and congenital nevi without cytologic atypia (step 1) may progress into dysplastic nevi with clear atypical histologic and cytologic features (step 2). Most of these lesions are stable, but a few may progress to a malignant melanoma that tends to grow outward along the radius of the plaque (step 3). Within the plaque, a nodule develops of fast-growing cells that expand in a vertical direction, invading the dermis and elevating the epidermis (step 4). Finally, the tumor metastasizes (step 5).
Adapted from Clark WH Jr, Elder DE, Guerry Dt, et al: A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol 15:1147–1165, 1984.
Self-Sufficiency in Growth Signals
Cells within normal tissues are largely instructed to grow by neighboring cells (paracrine signals) or via systemic (endocrine) signals. Similarly, cell to cell growth signaling also occurs in the vast majority of tumors. The immediate tumor cell environment (the stroma) contains resident nonmalignant cells such as parenchymal cells, epithelial cells, fibroblasts, and endothelial cells. In addition, most tumors are characterized by infiltrating immune cells such as lymphocytes, polymorphonuclear cells, mast cells, and macrophages. In some tumors, these cooperating cells may eventually transform themselves, coevolving with the tumor cells to sustain the growth of the latter. Finally, basement membranes form the extracellular matrix (ECM), which provides a scaffold for proliferation of fibroblast and endothelial cells. Together, tumor cells and stroma produce factors (autocrine and paracrine factors) that in cell-bound, matrix-bound, or soluble form directly or indirectly influence tumor development. Autocrine factors secreted by tumor cells promote growth of tumor cells but may also stimulate neighboring cells. In addition, tumor cells secrete paracrine factors that act on host cells or ECMs, generating a supportive microenvironment. For example, transforming growth factor-β (TGF)-β may induce angiogenesis, production of ECM molecules, and production of other cytokines by fibroblasts and endothelial cells. To state it simply, tumor growth is dependent on the response of tumor cells to paracrine and autocrine factors (Fig. 30-4, such as angiogenesis factors, growth factors, chemokines (polypeptide signaling molecules originally characterized by their ability to induce chemotaxis), cytokines, hormones, enzymes, and cytolytic factors, which may promote or reduce tumor growth (Table 30-3).
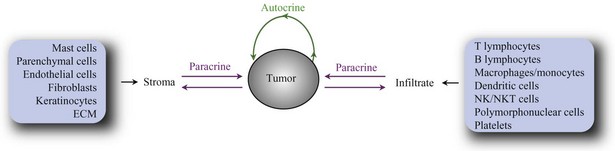
Table 30-3 Cells and Soluble Factors Affecting Tumor Development*
| CELLS | SOLUBLE FACTORS |
|---|---|
| Stroma | |
| Parenchymal cells | Growth factors, growth inhibitors, nutritional factors, hormones, degradative enzymes, cytokines, angiogenesis factors |
| Endothelial cells | |
| Fibroblasts | |
| Mast cells | |
| Extracellular matrix | |
| Keratinocytes | |
| Infiltrate | |
| T lymphocytes | Cytokines, chemokines, cytolytic factors, angiogenesis factors, growth (inhibitory) factors, degradative enzymes, cytostatic factors, antibodies |
| B lymphocytes | |
| Natural killer cells | |
| Natural killer T cells | |
| Macrophages, monocytes | |
| Dendritic cells | |
| Polymorphonuclear cells | |
| Platelets | |
| Tumor | |
| Chemokines, cytokines, angiogenesis factors, degradative enzymes, growth (inhibitory) factors |
* The list of cells and soluble factors is not meant to be complete but to illustrate the complexity of factors affecting tumor development.
During the evolution of a tumor, its responsiveness to growth signals changes. Paracrine growth mechanisms are dominant during the early development of tumor. Tumors become resistant to paracrine growth inhibitors and gain responsiveness to paracrine growth promoters. However, autocrine growth mechanisms become more prominent as tumors develop further. The observation that in late-stage tumors, metastatic tumor cells tend to spread more randomly throughout the body suggests that autocrine growth mechanisms may be more dominant than paracrine growth mechanisms. Advanced breast cancers, for example, lose hormone responsiveness. It is even possible for a tumor to grow completely autonomously (acrine state) and be independent of growth factors and inhibitors (Fig. 30-5).
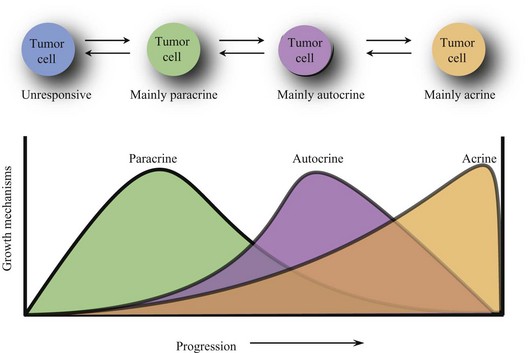
Insensitivity to Antigrowth Signals
Cell division is an ordered, tightly regulated process involving stimulatory and inhibitory signals. Thus, in addition to acquiring stimulatory growth signals, tumor cells need to overcome or neutralize growth inhibitory signals. These signals include soluble growth inhibitors and immobilized inhibitors embedded in the ECM and on the surfaces of neighboring cells. Similar to many of the stimulatory signals, the growth inhibitory signals are transduced by transmembrane receptors coupled to intracellular signaling pathways that target genes regulating the cell cycle. The cell cycle can be divided into an interphase and a mitotic (M) phase (Fig. 30-6).11 The interphase is further subdivided into two gap phases (G1 and G2), separated by a phase of DNA synthesis (S phase). The two gap phases involve crucial regulatory events that prepare the cell for DNA replication and mitosis.
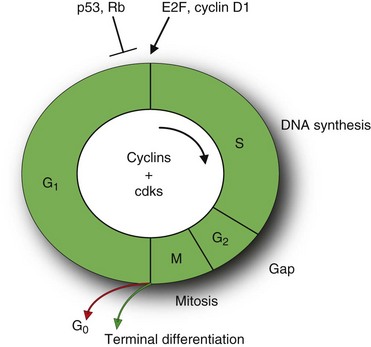
Central to cell cycle progression are the cyclin-dependent kinases (CDKs) that bind to the cyclin proteins. These proteins are regulated by numerous other proteins, including tumor suppressors and oncogenes that induce stimulatory or inhibitory signals. Antigrowth signals can block cell division by two distinct mechanisms. Cells may be forced to exit the cell cycle into a quiescent (G0) state (Fig. 30-6).
Alternatively, cells may be induced to enter a postmitotic state, usually associated with terminal differentiation. Much of the signaling pathways that enable normal cells to respond to antigrowth signals are associated with the cell cycle block, specifically with the components governing the restriction point in the G1 phase of the cell cycle. The restriction point marks the point between early and late G1 phase passage that represents an irreversible commitment to undergo one cell division. Cells monitor their external environment during this period and, on the basis of sensed signals, decide whether to proliferate, be quiescent, or enter into a postmitotic state. At the molecular level, many and perhaps all antiproliferative signals involve the retinoblastoma protein (pRb) and its two family members, p107 and p130.11 pRb is a key negative regulator at the restriction point. In quiescent cells, pRb is hypophosphorylated and blocks cell division by binding E2F transcription factors that control the expression of many genes essential for progression from the G1 into the S phase (see Fig. 30-6). In contrast, growth stimulatory signals induce phosphorylation of pRb that does not bind E2F factors and is considered functionally inactive. Similarly, disruption of the pRb pathway liberates E2Fs and thus allows cell proliferation, rendering cells insensitive to antigrowth factors that normally operate along this pathway to block advance through the G1 phase of the cell cycle. For example, TGF-β prevents the phosphorylation of pRb that inactivates pRb and thereby blocks advance through G1. In some tumors, such as breast, colon, liver, and pancreatic cancers, TGF-β responsiveness is lost through downregulation of TGF-β receptors or through the expression of mutant, dysfunctional receptors. In others, such as colon, lung, and liver cancers, the cytoplasmic Smad4 protein, which transduces signals from ligand-activated TGF-β receptors to downstream targets, may be eliminated through mutation of its encoding gene. Alternatively, in cervical carcinomas induced by human papillomavirus (HPV), the viral oncoprotein E7 binds pRb and thereby induces dissociation of E2F and subsequent transcription of genes necessary for cell cycle progression. In addition, cancer cells can also turn off the expression of integrins and other cell adhesion molecules that send antigrowth signals. In summary, the antigrowth signaling pathways converging onto Rb and the cell cycle are disrupted in most human cancers.
Evasion of Cell Death
The growth of tumors is determined by the ability of tumor cells to proliferate, offset by cell death. Most, if not all, types of tumors are characterized by defects in cell death–signaling pathways and are resistant to cell death. Cell death in tumors is caused primarily by programmed cell death, or apoptosis, which is the most common and well-defined form of cell death.12 Apoptosis is a physiologic cell suicide program essential for embryonic development, functioning of the immune system, and maintenance of tissue homeostasis. Apoptosis is characterized by disruption of membranes and chromosomal degradation in a matter of hours. The general apoptosis signaling pathway involves the release of cytochrome c from mitochondria that activates various caspases (a family of at least 10 proteases) in sequence (Fig. 30-7).
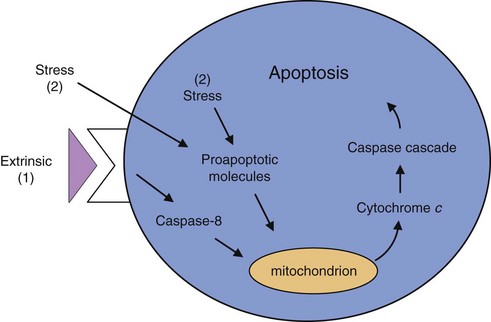
The concept that apoptosis forms a constraint to cancer was first put forth in 1972, when massive apoptosis was observed in cells populating rapidly growing, hormone-dependent tumors following hormone withdrawal.13 The discovery of the bcl-2 oncogene as having antiapoptotic activity led to the investigation of apoptosis in cancer at the molecular level.14 Bcl-2 promotes the formation of B cell lymphomas through a chromosomal translocation linking the bcl-2 gene to an immunoglobulin locus, which results in the constitutive activation of bcl2, driving lymphocyte survival. Further research has demonstrated that altering components of the apoptotic machinery allows a cell to resist death signals, providing it with a selective growth advantage. For example, functional inactivation of the tumor suppressor p53 is observed in more than 50% of human cancers. p53 is a key regulator of apoptosis by sensing DNA damage that cannot be repaired and subsequent activation of the apoptotic pathway. Other abnormalities, such as hypoxia and oncogene overexpression, are also channeled, in part via p53 to the apoptotic machinery, and fail to elicit apoptosis when p53 function is lost. Also, alterations in cell survival pathways can suppress or alter apoptosis. For example, the PI3 kinase-AKT pathway, which transmits antiapoptotic survival signals, is likely involved in inhibiting apoptosis in many human tumors. This signaling pathway can be activated by extracellular factors such as insulin-like growth factor 1 (IGF-1), IGF-2, or interleukin-3 (IL-3), intracellular signals from Ras, or loss of the pTEN tumor suppressor that negatively regulates the PI3 kinase–AKT pathway. A final example is the discovery of a nonsignaling decoy receptor for Fas ligand in a high fraction of lung and colon carcinoma cell lines. Expression of this decoy receptor dilutes the death signal mediated through Fas.
Nonapoptotic types of cell death include necrosis, autophagy, and mitotic catastrophe. Necrosis is normally induced by pathophysiologic conditions such as infection, inflammation, or ischemia. Necrosis is characterized by unregulated cell destruction. Autophagy, on the other hand, is characterized by the proteolysis of long-lived proteins and organelle components in lysosomes.15 Cells that undergo excessive autophagy undergo apoptosis. Autophagy is triggered by growth factor withdrawal, hypoxia, DNA damage, and differentiation and developmental triggers.12 Finally, aberrant mitosis caused by failure of the G2 checkpoint to block mitosis when DNA is damaged, can lead to cell death, known as mitotic catastrophe. The signaling pathways involved in these types of nonapoptotic cell death are less well defined compared with those that regulate apoptosis, but it is clear that defects in nonapoptotic cell death pathways have been linked to cancer. For example, amplification of the MDM2 oncogene, which negatively regulates expression of p53, results in inadequate expression of p53 and thereby loss of tumor suppressor function. Another example is the deletion of the autophagy-regulating gene becklin-1 in high percentages of ovarian, breast, and prostate cancers. In addition to cell death, cells can undergo permanent growth arrest, known as senescence, when repair of damaged DNA fails. Senescent cells lose their clonogenicity, but defects in the senescent program contribute to tumor development.
Limitless Replication Potential
With the exception of stem cells, activated lymphocytes, and germline cells, normal cells have a limited replicative potential. Stem cells give rise to progenitor cells that can progress through a certain number of doublings, with an increasing degree of differentiation. Fully differentiated cells do not have replicative potential. The number of doublings is controlled by telomeres, the ends of chromosomes that are composed of several thousand repeats of a short 6-bp sequence element.16 Telomeres prevent end to end chromosomal fusion. However, each DNA replication is associated with a loss of 50 to 100 base pairs of telomeric DNA from the ends of every chromosome. The progressive shortening of telomeres through successive cycles of replication eventually causes them to lose their ability to protect the ends of chromosomal DNA. When the critical length is bridged, the unprotected chromosomal ends participate in end to end chromosomal fusions, yielding a karyotype disarray that almost inevitably results in the death of the affected cell. Telomeric attrition is negated by the enzyme telomerase, which elongates telomeric DNA. Telomerase activity is high during embryonic development and in certain cell populations, such as adult stem cells. However, many tumors are characterized by elevated telomerase activity. Alternatively, telomeres are maintained through recombination-based interchromosomal exchanges of sequence information. Thus, by maintaining a telomere length above a critical threshold, the tumor cells have unlimited proliferative potential and are considered immortal.
Recently, evidence has been obtained for the existence of cancer stem cells or cancer-initiating cells that give rise to tissue-specific progenitor cells and phenotypically diverse cancer cells with limited replicative potential.17 Unlike mature, terminally differentiated tissue cells, cells with the capacity for self-renewal would live long enough for the stepwise accumulation of genetic mutations over time. Small populations of putative cancer stem cells have now been identified in many of the common cancers based on their ability to replicate, whereas most cancer cells have no or limited ability to proliferate. The most likely origin of cancer stem cells is normal adult stem cells that replace short-lived mature cells in tissues such as skin, gut, and blood. When normal stem cells divide, one of the daughter cells inherits stem cell capabilities, whereas the other cell is launched along the differentiation pathway. In cancer stem cells, the genes regulating self-renewal, such as Bmi-1, are overexpressed, thereby suppressing the default pathway of differentiation.
Sustained Angiogenesis
Based on the observation that many individuals who died of non–cancer-related causes had in situ tumors at the time of autopsy, physicians and scientists have concluded these microscopic tumors are in a dormant state. Tumor dormancy occurs because the body blocks the tumor from recruiting its own blood supply to provide tumor cells with the required oxygen and nutrients. The growth of new blood vessels, angiogenesis, is a highly regulated process to ensure supply to all cells in an organ. Surprisingly, the microscopic tumors lack the ability to induce angiogenesis, and only an estimated 1 in 600 acquire angiogenic activity. Research pioneered by Judah Folkman has demonstrated that naturally occurring endogenous angiogenesis inhibitors prevent tumors from expanding.18 The angiogenesis inhibitors keep the tumors in check by counterbalancing the angiogenic signals. These signals are mediated by soluble factors and their receptors on endothelial cells, as well as integrins and adhesion molecules mediating cell-matrix and cell-cell interactions. Angiogenic activity is induced by growth factors such as vascular endothelial growth factor (VEGF), basic and acidic fibroblast growth factor (FGF), and platelet-derived growth factor. Each binds to transmembrane tyrosine kinase receptors displayed primarily by endothelial cells connected to intracellular signaling pathways. Angiogenesis inhibitors are associated with specific tissues or circulate in the blood. The first inhibitor, interferon-α (IFN-α), was reported in 1980, and an additional 26 endogenous inhibitors have been identified since then. These include thrombospondin, tumstatin, canstatin, endostatin, and angiostatin.
The ability to induce and sustain angiogenesis seems to be acquired in a discrete step (or steps) during tumor development via a switch to the angiogenic phenotype.18,19 Tumors appear to activate the angiogenic switch by changing the balance between total angiogenic stimulation and total angiogenic inhibition.20 This usually occurs when the angiogenesis stimulators overwhelm the angiogenesis inhibitors. In some tumors, these changes may be linked. It is likely that such disruption in the angiogenic balance is under control of the genetic makeup of the individual tumor cell and its microenvironment. Angiogenesis inducers and inhibitors may be genetically controlled by tumor suppressor genes such as p53, whereas oncogenes such as ras may downregulate transcription of endogenous inhibitors or activate inducers. For example, bcl2 activation leads to significantly increased expression of VEGF and angiogenesis. Another dimension of regulation is through proteases, which can control the bioavailability of angiogenic activators and inhibitors. Thus, a variety of proteases can release bFGF stored in the ECM, whereas plasmin, a proangiogenic component of the clotting system, can cleave itself into an angiogenesis inhibitor form called angiostatin. Another angiogenesis inhibitor, endostatin, is an internal fragment of the basement membrane collagen XVIII. Finally, hypoxia and other metabolic stressors, mechanical stress from proliferating cells, or inflammatory immune responses can trigger angiogenesis. The coordinated expression of proangiogenic and antiangiogenic signaling molecules and their modulation by proteolysis appear to reflect the complex homeostatic regulation of normal tissue angiogenesis and vascular integrity. Different types of tumors use distinct molecular strategies to activate the angiogenic switch.
Tissue Invasion and Metastasis
Progressing tumors give rise to distant metastases that are the cause of 90% of human cancer deaths. The formation of tumor metastases is characterized by the detachment of some tumor cells from the primary tumor and infiltration into the bloodstream or lymphatics (intravasation). The reciprocal process occurs at other locations in the body (extravasation). Both intravasation and extravasation are characterized by changes in ECMs and their interactions with tumor cells. The cell-cell and cell-matrix interactions are mediated through cell adhesion molecules (CAMs), primarily by members of the immunoglobulin and calcium-dependent cadherin families,21 hyaluronan receptor CD44, selectins, and integrins,22 which link cells to ECM substrates. Studies have shown that the molecules mediating adhesion are also capable of signal transduction. As such, changes in expression of adhesion molecules will alter signaling pathways and, conversely, signaling molecules can directly affect the function of adhesion molecules in tumor cells.
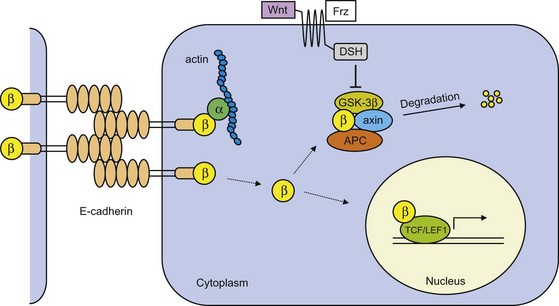
Epithelial (E)-cadherin is the prototype cadherin responsible for cell polarity and organization of epithelium. E-cadherin function is lost in most epithelial tumors during progression to tumor malignancy, and may in fact be a prerequisite for tumor cell invasion and metastasis formation. In normal cells, extracellular domains of E-cadherin on opposing cells couple and form cell-cell junctions (Fig. 30-8). The cytoplasmic cell-adhesion complex is linked to the actin cytoskeleton through catenins (α, β, γ). Mechanisms that include mutational inactivation of the E-cadherin or β-catenin genes, transcriptional repression, or proteases of the extracellular cadherin domain induce loss of E-cadherin function.21 This prevents catenins from binding and leads to their accumulation in the cytoplasm. Inactivation of nonsequestered β- and γ-catenin is dependent on the presence of the tumor suppressor gene APC and an inactive Wnt signaling pathway (Fig. 30-8). However, when APC function is lost, as is the case in many colon cancers or in case of Wnt activation, β-catenin is not degraded but instead translocates to the nucleus; here transcription is activated of genes involved in cell proliferation and tumor progression, such as c-myc, cyclin D1, CD44, and others.
Changes in expression of CAMs in the immunoglobulin superfamily also appear to play critical roles in the processes of invasion and metastasis.2,21 Neuronal (N)-CAM, for example, undergoes a switch in expression from a highly adhesive isoform to poorly adhesive (or even repulsive) forms in Wilms’ tumor, neuroblastoma, and small cell lung cancer. In invasive pancreatic cancer and colorectal cancers, the overall expression of N-CAM is reduced.
Changes in integrin expression are also evident in invasive and metastatic cells. For invading and metastasizing cells to be successful, they need to adapt to changing tissue microenvironments. This is accomplished through shifts in the spectrum of integrin α and β subunits displayed on the cell surface by the migrating cells. The large extracellular domain of integrins can bind to ECM molecules such as collagens, laminin, and fibronectin, ligands associated with vascular and coagulation physiology, such as thrombospondin and factor X, or with other CAMs. Each integrin molecule consists of an α subunit and β subunit, but a particular β subunit can dimerize with several different α subunits. These novel permutations result in different integrin subtypes—24 combinations have now been described—having distinct substrate preferences. Also, integrins may exhibit different specificities when expressed on different cell types. Thus carcinoma cells facilitate invasion by shifting their expression of integrins from those that favor ECM present in normal epithelium to other integrins that preferentially bind the degraded stromal components produced by extracellular proteases.2 For example, expression of α4β1, which binds fibronectin, correlates with the progression of melanoma. The changes are incompletely understood because of the large number of distinct integrin genes, by the even larger number of heterodimeric receptors resulting from combinatorial expression of various α and β receptor subunits, and by increasing evidence of complex signals emitted by the cytoplasmic domains of these receptors. Changes in integrin expression may also be essential for the expansion of the tumor stem cell compartment by inhibiting differentiation or apoptosis.23
Outgrowth at Preferred Sites
Another theory, the adhesion theory, proposes that endothelial cells lining the blood vessels in certain organs express adhesion molecules that bind tumor cells and permit extravasation. A third theory is that chemokines secreted by the target organ can enter the circulation and selectively attract tumor cells that express receptors for the chemokines. Evidence for the importance of chemokines in tumor progression has been obtained for breast cancer cells preferentially metastasizing in bone marrow, liver, lymph nodes, and lung.24 These organs were found to secrete CXCL12, which is the ligand for the chemokine receptor, CXCR4, enriched on breast cancer cells compared with normal breast epithelial cells. A similar phenomenon was observed for melanoma cells that were found to express elevated levels of the receptors CXCR4, CCR7, and CCR10 compared with normal melanocytes. Lymph nodes, lung, liver, bone marrow, and skin express the highest levels of the ligands for these receptors and are the preferred sites for metastatic spread of melanomas. Because chemokines are now known to affect angiogenesis and expression of cytokines, adhesion molecules, and proteases, in addition to inducing migration, it appears that chemokines and their receptors play an essential role in the successful outgrowth of tumors at preferential sites.
Central to the mechanisms dictating metastatic predisposition are bone marrow–derived progenitor cells expressing the VEGF receptor 1 (VEGFR1) and VLA-4, which are prompted by the primary tumor to establish premetastatic niches before arrival of metastatic tumor cells.25 Tumor-secreted humoral factors induce fibronectin (a VLA-4 ligand) expression on fibroblasts and fibroblast-like cells in specific distant organs. Simultaneously, the VEGFR1+, VLA-4+ cells leave the bone marrow and migrate to the premetastatic niche, where they form cellular clusters that permit the development of metastases.
Immunosurveillance and Immunoediting
Immunosurveillance
In the early 1900s, Paul Ehrlich proposed that the frequency of cancerous transformations would be very high if it were not for the defense system of the host. This concept was later substantiated in the 1950s and 1960s, and the term immunosurveillance was introduced by Burnet in 1970. Burnet hypothesized that the development of T lymphocyte–mediated immunity during evolution was specific for the elimination of transformed cells. He further proposed that there is a continuous surveillance of the body for transformed cells—hence, the term immunosurveillance. During the subsequent years, experiments in immunosuppressed and immunodeficient mice demonstrated that T cell–mediated immunity provides protection against virally induced tumors. However, no conclusive evidence was obtained for the immunosurveillance of cancer. More recent discoveries have made it clear that the earlier studies were performed in mice erroneously assumed to be immunodeficient. When tested in truly immunoincompetent mice, evidence for immune surveillance of cancer was obtained; immunodeficient mice were significantly more susceptible to the formation of chemically induced tumors and spontaneous tumors than immunocompetent mice.1 This suggests that the unmanipulated immune system is capable of recognizing and eliminating primary tumors.
Does immunosurveillance of cancer exist in humans? Evaluation of long-term studies in transplant patients who were immunosuppressed and patients with immunodeficiencies has shown an increased incidence of virally induced tumors, such as non-Hodgkin’s lymphoma, Kaposi’s sarcoma, and carcinomas of the genitourinary and anogenital regions. However, they also showed a higher incidence of tumors with no apparent viral cause, such as malignant melanoma, lung cancer, pancreatic cancer, colon cancer, and kidney cancer. More conclusive were observations from patients with paraneoplastic neurologic degenerations (PNDs).26 These patients develop autoimmune neurologic disease in discrete regions of the nervous system, mediated through antibodies and cytotoxic T cells against neuronal antigens. Clinical examination reveals systemic malignancies, usually breast or ovarian adenocarcinoma or small cell lung cancer that are generally small, show limited spread, and are sensitive to treatment. Importantly, the presence of antineuronal T cells and antibodies in all PND patients studied is associated with clinical and pathologic evidence of suppression of tumor growth. Some cancer patients mount a PND immune response but do not develop neurologic disease. These patients have smaller tumors and longer survival than those without such immune responses. Finally, extensive studies on immune infiltrate in primary human cancers have established that the presence of memory T cells, particularly of the T helper 1 subtype, and cytotoxic T cells are prognostic factors for disease-free and overall survival at all stages of clinical disease.27 The data from mouse and human studies combined suggest that immunosurveillance of cancer does exist, mediated through immune cells and soluble factors. Although the immune system may eliminate most transformed cells, some cells manage to escape and may develop into tumors.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


