31 Troubleshooting of Biventricular Devices
 Electromechanical Events and Cardiac Pump Function
Electromechanical Events and Cardiac Pump Function
Consequences of Uncoupling at Various Levels
Uncoupling at the Atrial Level
The right atrium and left atrium are activated nearly simultaneously (within 50-80 milliseconds) during sinus rhythm. Preferential sites of interatrial conduction exist at the posterior-superior interatrial septum (Bachmann’s bundle region), fossa ovalis, and coronary sinus ostium.1,2 Significant interatrial conduction delays (up to 200 msec or greater) can occur in myopathic atria. Similar conduction delays can be also be induced, or exacerbated, by right atrial (RA) pacing. Delayed left atrial (LA) contraction can disrupt optimal left-sided atrioventricular (AV) coupling. Severe atrial decoupling and delayed LA contraction reverses the left-sided AV contraction sequence, resulting in atrial transport block.3 This causes increased LA pressures, retrograde flow in the pulmonary veins, and counterphysiologic neurohormonal responses termed pseudo–pacemaker syndrome.4–6
Uncoupling at the Atrioventricular Level
Optimal AV coupling contributes to ventricular pump function through Starling’s law. The normal intrinsic atrioventricular interval (iAVI) results in atrial contraction just before the preejection (isovolumetric) period of ventricular contraction that maximizes left ventricular (LV) filling (LV end-diastolic pressure, or preload) and cardiac output. Appropriately timed AV coupling maintains a low mean atrial pressure, increases LV filling time, maximizes preload and pump function (Starling mechanism), and positions the mitral valve for closure during the isovolumetric phase of ventricular systole (Fig. 31-1).
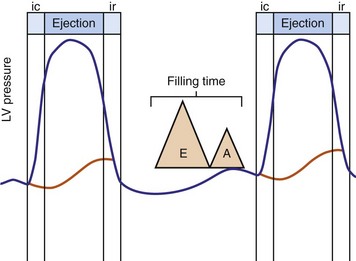
Delayed AV coupling resulting from AV conduction delay displaces atrial contraction earlier in diastole (Fig. 31-2). Atrial contraction occurs during ventricular filling, which shortens diastolic filling time, and in extreme situations, occurs immediately after or coincident with the preceding ventricular contraction. This situation is aggravated by ventricular conduction delay, which increases ventricular ejection time and delays diastolic filling, increasing the probability of collision with atrial contraction. Atrial contraction before completion of venous return reduces preload stretching of the left ventricle, which reduces ventricular volume and contractile force. Delayed AV coupling reduces diastolic filling time and may result in diastolic mitral regurgitation when elevated LV end-diastolic pressure exceeds LA pressure. Partial closure of the mitral valve may also occur, further shortening diastolic filling time.
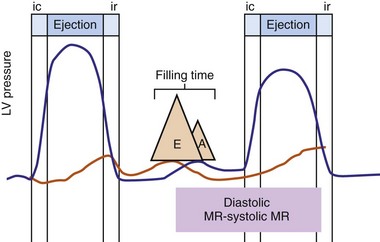
Figure 31-2 Schematic of hemodynamic effects of AV decoupling on LV pump function.
Same arrangement as in Figure 31-1. Atrial contraction truncates passive diastolic filling (E-A fusion). A late diastolic left ventricular–left atrial (LV-LA) pressure gradient before mitral valve closure results in diastolic mitral regurgitation. LV filling is reduced and pump function declines (reduced LV pressure during ejection).
Delayed AV coupling may be worsened or induced by atrial pacing. This is a consequence of atrial capture latency, increased atrial conduction time resulting from altered activation, and increased AV nodal conduction time from altered atrial input. These effects of atrial pacing are enhanced by increased heart rates during atrial pacing and autonomic changes during sleep.7
Severe AV decoupling coexists with ventriculoatrial (VA) coupling. In extreme conditions LA contraction occurs immediately after, within, or preceding the LV contraction, causing reversal of the left-sided AV contraction sequence,3 which initiates counterphysiologic circulatory and neurohumoral reflexes. These derangements (pseudo–pacemaker syndrome) have been rarely described during marked first-degree AV block and atrial pacing.4 The identical pathophysiology occurs during AV synchronous ventricular pacing when LV contraction timing is too early, resulting in LA transport block (Fig. 31-3). This situation is corrected by proper timing of ventricular contraction with pacing (see Atrioventricular Optimization).
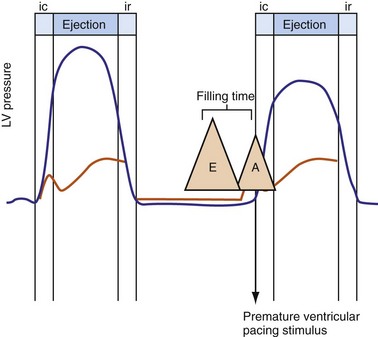
Figure 31-3 Hemodynamic effects of short A-V interval on LV pump function.
Same arrangement as in Figures 31-1 and 31-2. Ventricular contraction triggered by premature pacing stimulus (arrow) truncates atrial emptying (atrial transport block). Atrial pressure increases, LV filling decreases, and pump function declines (reduced LV pressure during ejection).
Ventricular Level
Normal LV electrical activation is rapid and homogeneous with minimal temporal dispersion throughout the wall.8 This elicits a synchronous mechanical activation and ventricular contraction.9 The resulting coordinated myocardial segment activation maximizes ventricular pump function. Wiggers10 established the linkage between pacing-induced alterations in ventricular conduction and pump function with two seminal observations in 1925: (1) pacing at virtually any ventricular site disturbs the natural pattern of activation and contraction, because the evoked electrical wavefront propagates slowly through ventricular myocardium rather than through the His-Purkinje system, and (2) altered ventricular activation causes an immediate reduction in pump function, and some sites are worse than others (site selectivity). In general, right ventricular (RV) pacing sites appear to be more detrimental than LV pacing sites, and the right ventricular apex (RVA) is among the worst sites within the right ventricle.11
Optimal interventricular and intraventricular coupling is more important than AV coupling for maximum ventricular pumping function.12,13 RVA pacing and left bundle branch block (LBBB) both induce delays in transseptal and intraventricular conduction.14 Consequently, the hemodynamic effects of altered ventricular activation during RVA pacing and LBBB are comparable. These effects can be attributed to disturbed interventricular as well as intraventricular coupling. RVA pacing also disturbs RV activation as a result of slow intramyocardial conduction.
Interventricular decoupling refers to a sequential RV-LV activation delay. In LBBB the earliest ventricular depolarization is recorded over the anterior RV surface and generally latest at the posterior or posterolateral basal LV surface.15–20 Interventricular dyssynchrony can be quantified by the time delay between the upslopes of LV and RV systolic pressure,12 as well as time delay between opening of the pulmonic and aortic valves.21 Similar changes occur during RV pacing. This disruption to ventricular interdependence is a determinant of paradoxical septal motion. Presumably this reduction in ventricular septal contribution to LV ejection is an important factor in the reduction of pump function during LBBB.12,22
Delayed intraventricular activation is presumably the most important determinant of reduced pump function. In RVA pacing and LBBB, electrical activation starts in the interventricular septum, with the posterior or posterolateral basal LV wall activated more than 100 msec later. Such considerable intraventricular delay during RVA pacing and LBBB pacing is caused by slow spread of the depolarization wavefront through the working myocardium rather than through the Purkinje system. Slow intramyocardial conduction during RVA pacing prolongs QRS duration (QRSd) from the normal values of about 80 msec to values of 140 msec in otherwise normal hearts, to about 200 msec in infarcted left ventricles.14 Furthermore, the delay in LV activation relative to QRSd during RVA pacing is greater in failing ventricles.23
Left bundle branch block is a complex electrical disease resulting from LV conduction delay at multiple anatomic levels (fixed or functional) and is exhibited differently according to substrate characterization (ischemic vs. nonischemic dilated cardiomyopathy [DCM]). Early endocardial activation mapping studies of LBBB concluded that conduction delay resided entirely within the ventricular septum, and that LV endocardial activation was rapid after single-point breakthrough in nonischemic DCM, whereas activation was slowed across regions of scar throughout the left ventricle in ischemic DCM.15 In both situations the posterior or posterolateral basal left ventricle is the latest activated region.
Higher-resolution endocardial and epicardial mapping studies have revealed that LV activation during LBBB, despite a similar surface electrocardiographic appearance, is considerably heterogeneous. At least three patterns of delayed LV activation have been characterized: (1) transseptal delay with or without LV endocardial delay, (2) normal transseptal conduction with slow conduction velocities in peri-infarct regions and globally slow in nonischemic DCM, and (3) slowed U-shaped activation around a line of functional block on the anterior wall.16–20 Generally, QRSd of 120 to 150 msec indicates delay confined specifically to the specialized conduction system, whereas QRSd greater than 150 msec usually indicates additional conduction delay in diseased myocardium.
The pattern of transseptal activation delay varies and influences LV endocardial activation. Significantly prolonged transseptal conduction times through a diseased left bundle branch result in midseptal LV or septoapical breakthroughs and unidirectional wavefront propagation throughout the left ventricle. Rapid transseptal conduction times typify conduction through septal branches of the His-Purkinje system, with basal breakthroughs from the anterior or posterior fascicle and bidirectional wavefront propagation from base to apex and high septum. Breakthrough from both fascicles results in a double wavefront that fuses on the posterolateral wall. Single-site breakthrough from the high septum caused by right-to-left muscular conduction (no conduction system present) results in a single wavefront that propagates from base to apex.16 The pattern of septal activation may affect ventricular mechanics differently. High septal activation may result in simultaneous RV and LV activation in opposite directions, and papillary muscle activation may be influenced by the earliest site of LV activation.16,20,24,25
Anterior locations of the line of functional block are characterized by a U-shaped LV activation pattern,17–19 more prolonged QRSd (>150 msec), and longer time to LV breakthrough.18 Late activation of the posterior or posterolateral basal left ventricle occurred by wavefront propagation around the line of block using the apical or inferior LV walls. Lateral locations of the line of block are characterized by less prolonged QRSd (<150 msec) and shorter time to LV breakthrough. Pacing maneuvers shift either line of block, indicating their functional nature. Noninvasive mapping of epicardial activation using body surface potentials extended these observations and demonstrated that electrical activation patterns in LBBB are highly heterogeneous and unpredictable.20 Lines of conduction block do not correlate with regions of wall motion abnormality or scarring. Some lines of block arose only during pacing and were site dependent (functional), whereas other lines of block could not be manipulated with pacing maneuvers, indicating these were caused by slow or absent conduction (fixed). Latest activation most often occurred in the posterior or posterolateral basal wall, but was also observed in the anterior and inferior walls in some patients.
Effects of Asynchrony on Left Ventricular Mechanics and Structure
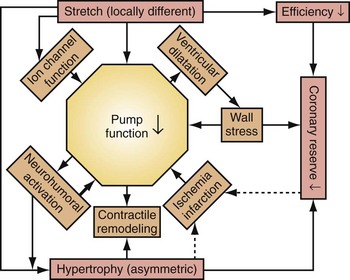
Regions that are activated early also start to contract early. Accordingly, in RVA pacing and LBBB, earliest and latest sites of segmental LV activation correlate well with time to peak systolic velocity by Doppler imaging19 and strain by tagged magnetic resonance imaging (MRI),9,26–28 providing evidence that ventricular conduction delay is responsible for heterogeneous mechanical performance (asynchrony). The mechanical effect of asynchronous electrical activation is dramatic, because the various regions differ not only in the time of onset of contraction, but also in the pattern of contraction (Fig. 31-4). Contraction of early-activated myocardium is energetically inefficient because LV pressure is low and ejection has not begun. Instead, stretching of remote regions not yet activated absorbs the energy generated by the early-activated regions. This stretching further delays shortening of these late activation regions and increases their force of local contraction by virtue of the Frank-Starling mechanism (locally enhanced preload). Vigorous late systolic contraction at delayed sites occurs against high LV cavity pressures (locally enhanced afterload) and imposes loading on the earlier activated regions, which now undergo systolic paradoxical stretch.29 This reciprocated stretching of regions within the LV wall causes a less effective and energetically efficient contraction.30
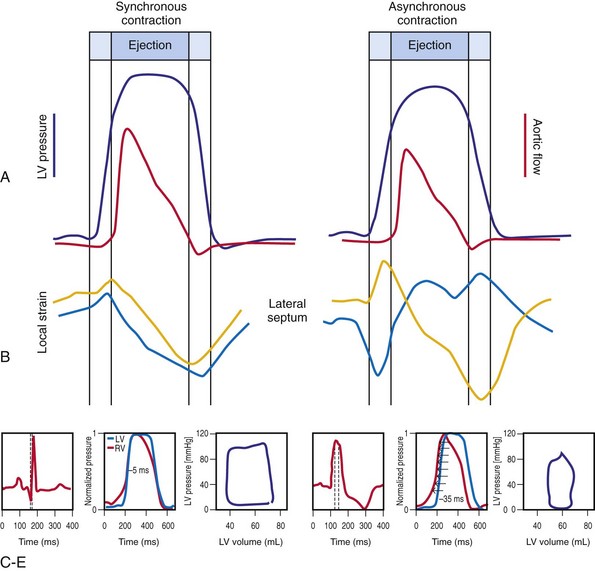
The hemodynamic consequences of the asynchronous LV contraction are reductions in contractility and relaxation. These changes occur immediately on initiating RVA pacing in humans and animals,11,31 and induction of LBBB in animals12 (see Fig. 31-4). The loss of pump function is indicated by decreases in stroke volume, stroke work, and slower rate of rise of LV pressure (Fig. 31-5). Moreover, the LV end-systolic pressure-volume relationship shifts rightward, indicating that the left ventricle operates at a larger volume in order to recruit the Frank-Starling mechanism.11 Premature relaxation in early-activated regions, and delayed contraction in others also causes abnormal relaxation,11 as expressed as slower rate of fall of LV pressure, increase in the relaxation time constant tau, and decrease of E-wave velocity amplitude on Doppler echocardiograms.32 These changes also lead to prolongation of the isovolumetric contraction and relaxation times, which are characteristic for asynchronous hearts.33 The prolongation of the isovolumetric times occurs mainly at the expense of the diastolic filling time, leading to reduced preload.
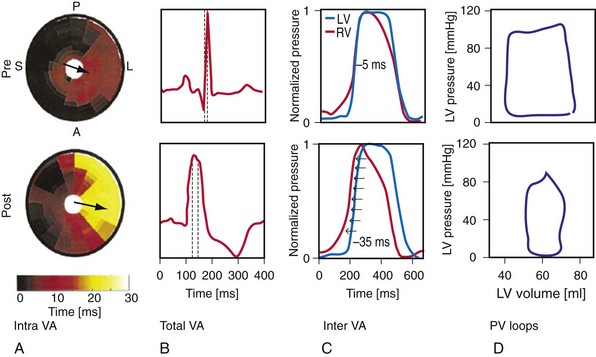
Figure 31-5 Left bundle branch block (LBBB)–induced asynchrony causes immediate reduction in LV contractility.
(Modified from Vernooy K et al: Left bundle branch block induces ventricular remodeling and functional septal hypoperfusion. Eur Heart J 26:91-98, 2005.)
The redistribution of the mechanical load within the ventricular wall also leads to reduction of regional myocardial perfusion and oxygen consumption near the RVA pacing site30,34,35 and in the septum during LBBB.36,37 Such perfusion defects and wall motion abnormalities have been demonstrated in up to 70% of patients with angiographically normal coronary arteries exposed to chronic RVA pacing.38–41 These defects are reversible on cessation of RVA pacing39,41 and after biventricular pacing.36,37,42 Accordingly, such perfusion “deficits” do not necessarily indicate coronary heart disease, because they may simply express the regional differences in myocardial workload.
Similar to the situation after myocardial infarction, acute loss of pump function initiates compensatory responses (Figs. 31-6 and 31-7). Some of these responses, after a certain time and or degree of asynchrony, may result in further impairment of pump function and clinical heart failure. There are various triggers for these “remodeling” processes. As for other conditions of hemodynamic overload, RVA pacing and LBBB lead to stimulation of the sympathetic system, resulting in elevated myocardial catecholamine levels,43 and activation of the renin-angiotensin-aldosterone system. Regional differences in stretch and mechanical load heterogeneity are most likely important stimuli for remodeling processes. Chronic asynchronous LV activation results in regional and global structural changes indicated by asymmetric hypertrophy, increased end-diastolic volume, and reduced ejection fraction36,44 (see Fig. 31-6), as well as locally different molecular abnormalities, including reductions in sarcoplasmic reticulum calcium-ATPase and phospholamban.45 Even stronger regional differences in gene expression are found in failing hearts with conduction abnormalities.46 Dystrophic calcifications, disorganized mitochondria, and myofibrillar cellular disarray47 have been described with RVA pacing, especially in immature hearts.
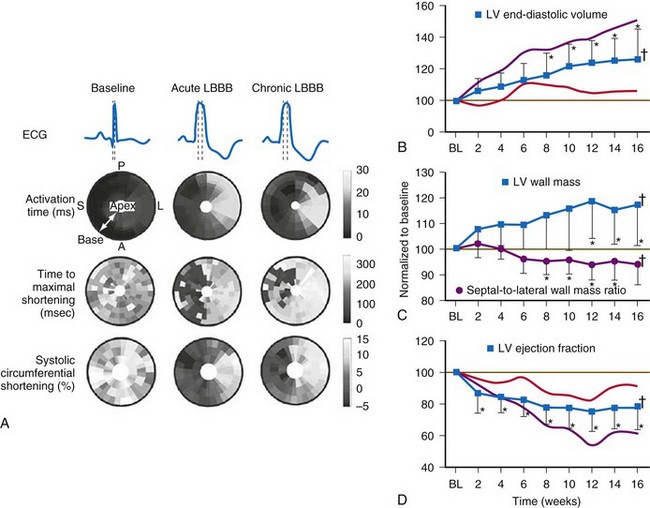
Figure 31-6 Left bundle branch block (LBBB).
(Modified from Vernooy K et al: Left bundle branch block induces ventricular remodeling and functional septal hypoperfusion. Eur Heart J 26:91-98, 2005.)
Early signs of cellular and molecular adaptation to disturbed ventricular activation are manifest as reduction in ejection fraction (EF) within the first week of RVA pacing. In patients with normal EF, Nahlawi et al.31 showed an immediate drop in EF of about 7%, followed by another 7% drop during the subsequent week. Suppression of RVA pacing returned EF to baseline values, but only after several days.48 Also, ventricular repolarization abnormalities have been observed within hours of RVA pacing. Alterations in potassium and calcium channels likely play a role in these phenomena.
Mechanistic Basis for Development of Asynchronous Heart Failure Caused by Ventricular Conduction Delay
As mentioned, synchronous activation consistently leads to acute and long-term adverse effects on cardiac pump function, which create a vicious cycle of deterioration. Accordingly, the pacing-induced ventricular conduction delay has been linked to increased risks of atrial fibrillation, heart failure, ventricular tachyarrhythmia, and death in large, randomized clinical trials of pacemakers and ICDs.49–56 Similar risks have been reported for LBBB and cardiac morbidity and mortality.57
Multiple factors may contribute to heart failure associated with ventricular asynchrony. At least three candidate factors are readily identified: (1) reduced pump function caused by asynchronous contraction (dyssynchrony), (2) adverse remodeling caused by long-term dyssynchrony, (3) left-sided AV desynchronization, and (4) functional mitral regurgitation (fMR). The first three factors are discussed earlier. The fMR frequently accompanies pacing-induced or spontaneous asynchronous heart failure and has multiple mechanisms.24,25,58,59 Assuming the anterior mitral leaflet is structurally normal, the immediate cause of fMR is delayed activation of the LV posterior “apparatus” caused by ventricular conduction delay.25,60 This apparatus consists of the posterior leaflet and annulus, the LV summit it sits on, the chordae, the papillary muscles, and the LV wall from which they emanate. Rapid activation of the posterior apparatus requires normal ventricular conduction.
Early closure of the mitral valve is arguably the most important accomplishment of the entire conduction system because it facilitates Starling’s law; that is, it permits isovolumetric systole to occur when the ventricle is as full as it can be given the filling conditions. Delayed ventricular conduction prevents mitral valve coaptation and induces fMR during isovolumetric contraction. Ventricular conduction delay instantaneously reduces the transmitral pressure gradient (systolic LV pressure–LA pressure difference) or mitral valve closing force, by reducing contractility (↓LV dP/dt). Chronically, volumetric remodeling contributes to fMR by further reductions in contractility (closing forces) and increasing papillary muscle tethering forces59,61,62 (Fig. 31-8). Tethering strongly depends on alterations in ventricular shape because the tethering forces that act on the mitral leaflets are higher in dilated, more spherical ventricles. Ventricular dilation and increased chamber sphericity increase the distance between the papillary muscles to the enlarged mitral annulus as well as to each other, restricting leaflet motion and increasing the force needed for effective mitral valve closure. Under these conditions, the mitral regurgitant orifice area will be largely determined by the phasic changes in transmitral pressure.
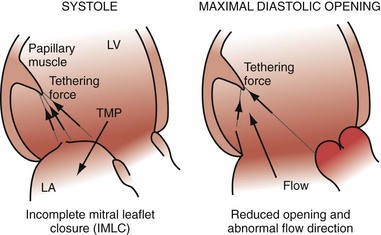
Figure 31-8 Effects of augmented mitral valve leaflet tethering in dilated cardiomyopathy.
(Modified from Otsuji Y, et al: Restricted diastolic opening of the mitral leaflets in patients with left ventricular dysfunction: evidence for increased valve tethering. J Am Coll Cardiol 32:398-404, 1998.)
There is some intriguing evidence that LBBB may be the primary cause of DCM in some patients. Progressive LV enlargement after the emergence of LBBB unaccompanied by any other explanation has been documented in case series.63 Furthermore, complete recovery of ventricular function shortly after initiation of biventricular pacing in “hyperresponders” suggests that LBBB may be a primary and reversible trigger of DCM in some patients.64,65
Several studies also showed that LBBB is associated with increased mortality, with greater impact in sicker patient populations. The relative risk associated with the presence of LBBB varied between 1.5 and 2.0, even after adjustment for covariates.57 The only study rejecting the association of LBBB with increased mortality drew this conclusion after adjustments for LV EF as a covariable.66 However, the conclusion that LBBB is not an independent predictor of mortality after the adjustment for EF can be understood because LBBB directly reduces EF because of its acute and chronic effect on LV pump function (see earlier).
Mechanisms of Cardiac Resynchronization Therapy
Fixed and functional disruptions to normal electromechanical ordering caused by conduction delay are targeted for electromechanical reconstitution of pump function. CRT has two immediate effects on electromechanical ordering: ventricular activation sequence and chamber timing. The basis of this therapeutic strategy is to restore coordinated contraction by minimizing the conduction delay, thereby restoring interventricular and intraventricular coupling. A second-order effect is the reduction of left-sided AV decoupling, which may improve diastolic performance and thus LV pumping function. As part of this effect, fMR may be reduced. These acute beneficial hemodynamic effects may lead to even more beneficial long-term effects, because many adverse molecular and cellular derangements elicited by chronic RVA pacing or LBBB appear to be reversible. This property of CRT is remarkable; many pharmacologic therapies tend to lose effectiveness over time, whereas the beneficial effects of CRT are maintained for many years. The best explanation of the lasting benefit of CRT is that it largely restores the normal electromechanical coupling of the heart with LBBB. This idea is supported by studies in the canine models in which CRT abolished all adverse effects of LBBB.42 Thus, CRT may be “curative” of LBBB-induced ventricular “desynchronapathy” in some situations.
Improved Atrioventricular Mechanics from Optimal AV Resynchronization
When LV contraction timing is delayed, even when AV conduction times are normal, the hemodynamic consequences are similar to a prolonged AV interval, as shown in Figure 31-2. Left-sided AV decoupling, due to LV conduction delay and/or AV conduction delay, is corrected with LV pacing. Optimal AV timing improves LV pump function, but the primary mechanism of improved ventricular mechanics during CRT is reduction in ventricular conduction delay. AV timing considerations during CRT are considered in detail later.
Improved Ventricular Mechanics from Reduced Ventricular Conduction Delay
Reduction of intraventricular delay during atrial-synchronous LV or biventricular pacing has an immediate positive effect on ventricular mechanics and is the dominant therapy effect of CRT. Instantaneous improvement in pump function is indicated by increases in LV +dP/dtmax, stroke volume, stroke work, arterial pulse pressure and peak systolic pressure,67,68 and reduced end-systolic volume (Fig. 31-9). Moreover, unlike the inotropic effects of dobutamine, systolic augmentation with ventricular resynchronization increases efficiency of conversion of myocardial oxygen consumption to mechanical work.69 This positive contractile response demonstrates a modestly positive correlation with increasing baseline QRSd67–69 and is strongly correlated with baseline mechanical dyssynchrony.69,70 Conversely, neither improved contractility nor reverse volumetric remodeling requires QRS narrowing during LV or biventricular pacing.68,71,72 However, unlike the inotropic effects of dobutamine, systolic augmentation with ventricular resynchronization does not increase detrimental myocardial oxygen consumption. These acute improvements in ventricular mechanics are maintained chronically and are accompanied by reverse volumetric remodeling of the LV (see later).73 Abrupt discontinuation of LV pacing results in immediate reduction in indices of improved contractility and regression of reverse remodeling over several weeks,73,74 as well as recurrence of fMR.25
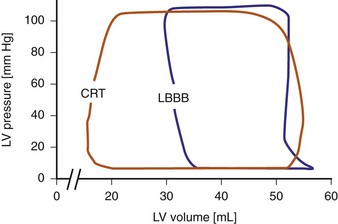
Figure 31-9 Pressure-volume loops during LBBB and LV pacing.
(Modified from Verbeek XA, Vernooy K, Peschar M: Intra-ventricular resynchronization for optimal left ventricular function during pacing in experimental left bundle branch block. J Am Coll Cardiol 42:558-567, 2003.)
Maximally effective ventricular resynchronization occurs at the midpoint of the interventricular (V-V) interval (Fig. 31-10), which is the conduction time from RV to LV pacing site during native activation. A single optimum AV delay will achieve maximally effective ventricular resynchronization by advancing LV activation sufficiently to minimize the V-V interval. The AV delay and the V-V interval have an interactive relationship. The effect of AV delay on acute hemodynamic response to CRT is best understood in terms of the effect on ventricular resynchronization during biventricular (BiV) or left univentricular pacing (Fig. 31-11). During LBBB, the right ventricle is activated by right bundle branch conduction (RBBc) before the left ventricle, and the time difference between RV and LV activation is the interventricular conduction delay.13 BiV pacing at short atrioventricular delay (AVD) results in complete replacement of intrinsic activation, allowing complete control of chamber timing with sequential ventricular stimulation. Intermediate AVDs result in fusion among RBBc, RV paced activation, and LV paced activation. Long AVDs result in “pure” fusion between RBBc and LV paced activation since LV preexcitation is still possible because of interventricular conduction delay.13,75,76 Modification of the AVD alone can therefore achieve (1) BiV paced fusion, (2) BiV paced fusion with sequential ventricular stimulation (LV first), and (3) “pure” RBBc-LV pacing fusion. Therefore, LV preexcitation is possible across a range of AVDs because LV time is delayed relative to RV time. RV pacing is not necessary to achieve fusion, and AVD can be used to time the relative prematurity of LV preexcitation. At very short AVDs, this requires sequential VV timing. At very long AVDs, this occurs spontaneously because of the native V-V interval.
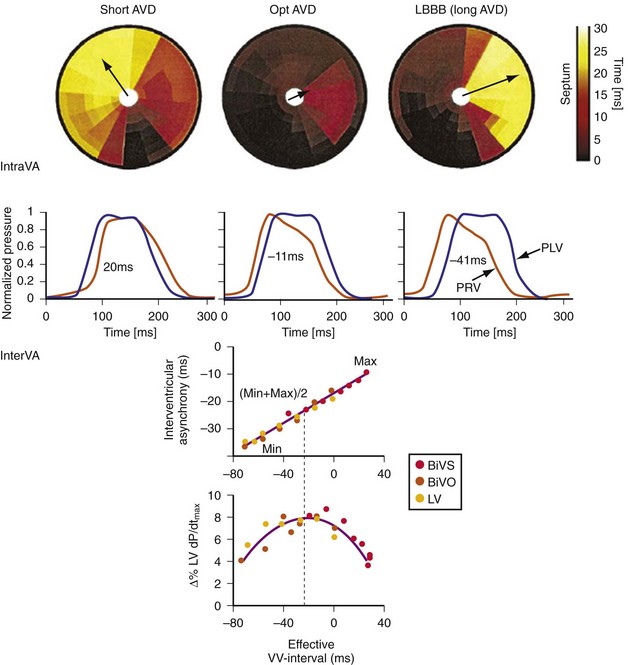
Figure 31-10 Relationship between intraVA and interVA under various LV pacing conditions.
(Modified from Verbeek XA, Vernooy K, Peschar M: Intra-ventricular resynchronization for optimal left ventricular function during pacing in experimental left bundle branch block. J Am Coll Cardiol 42:558-567, 2003; and Vernooy K, et al: Calculation of effective VV interval facilitates optimization of AV delay and VV interval in cardiac resynchronization therapy. Heart Rhythm 4:75-82, 2006.)
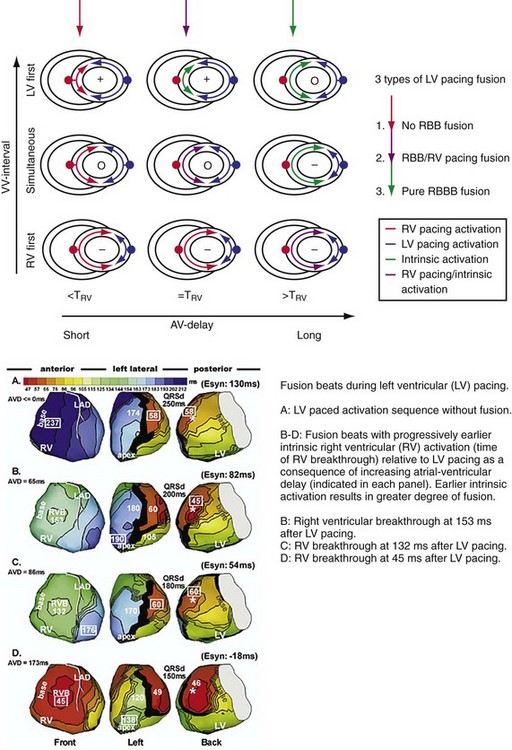
Figure 31-11 Top, Interaction between atrioventricular delay (AVD) and V-V interval to determine LV activation fusion. T-RV is time to intrinsic RV activation (RA sense to RV sense, or P-R interval). During short AVD pacing (AVD < T-RV), biventricular pacing completely replaces native activation (surrogate fusion), and relative timing of ventricular activation is determined by sequential ventricular stimulation (RV first, simultaneous, LV first). When AVD = T-RV, RV pacing occurs simultaneously with intrinsic conduction, resulting in a combination of RV pacing and intrinsic conduction, whereas left ventricle is still activated earlier than would have occurred due to LBBB. When AVD > T-RV, LV-only pacing occurs with true fusion of native and paced activation. Bottom, Epicardial isochrone maps are shown during LV-only pacing at increasing pacemaker A-V intervals (pAVIs). Activation times relative to QRS onset are color-coded with red earliest and blue latest; framed numbers indicate earliest and latest activated sites. Asterisk, LV stimulation site. Black lines indicate line/region of conduction block, which influences activation wavefront propagation (see Fig. 31-77). A, pAVI = 0 msec. Note sequential ventricular activation (LV→RV): LV is activated earliest, and latest activation occurs at posterobasal right ventricle. B to C, Progressive fusion of paced LV activation with intrinsic LV activation via RBB as pAVI is increased from 65 to 86 msec. Note also progressively early RV activation from RBB conduction. D, Maximum ventricular activation wavefront fusion at pAVI = 173 msec. Note right and left ventricles are simultaneously activated (Esyn, electrical synchrony, = −18 msec).
(Modified from Jia P et al: Electrocardiographic imaging of cardiac resynchronization therapy in heart failure: observation of variable electrophysiologic responses. Heart Rhythm 3:296-310, 2006.)
Early acute hemodynamic studies demonstrated that the relationship between AVD and LV dP/dtmax in patients with an acute improvement in contractile response to simultaneous BiV pacing is positive and unimodal, with a peak effect at approximately 50% of the native P-R interval, resulting in complete replacement of native ventricular conduction with paced activation67,68,77 (Fig. 31-12). This dose-response relationship between AVD and acute contractile response shown is interpreted as follows. The magnitude of the acute contractile response depends on the magnitude of baseline asynchrony (ventricular conduction delay, QRSd) and the AVD, which titrates how much intrinsic ventricular activation is replaced by pacing activation. Patients with greater ventricular conduction delay (QRS >150 msec) benefit from near-complete replacement of intrinsic activation with BiV or LV pacing (i.e., short AVDs). Patients with less ventricular conduction delay (QRS <150 msec) benefit from limited replacement of intrinsic activation (e.g., long AVDs). Typically, programmed short AVDs of 100 to 120 msec, although near-optimal for some patients, may result in decreased contractile function in patients with less baseline asynchrony (e.g., QRS <150 msec) because even BiV pacing induces asynchrony.9 LV dP/dtmax increased 15% to 45% across a narrow range of AVDs and declined at very short AVDs (truncated filling times) or very long AVDs (inadequate correction of LV conduction delay). This maximum improvement in pump function (LV +dP/dtmax and stroke volume) occurs at minimal intraventricular asynchrony and unchanged LV end-diastolic pressure (preload).13 This indicates that the acute contractile response is explained by ventricular resynchronization, not by better filling, and that the dose of resynchronization is titrated by the AVD.
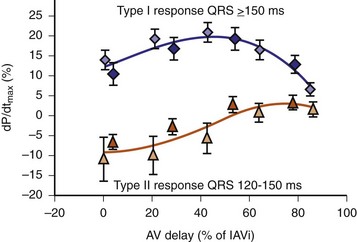
The best ventricular resynchronization occurs when the wavefronts from the RV pacing site (or originating from RBBc) and the LV pacing site collide halfway.78 Experimental evidence suggests that optimal hemodynamic effect can be achieved using LV pacing at certain AVDs as well as using simultaneous and sequential BiV pacing because fusion of electrical wavefronts can be achieved by either approach13,79 (see Fig. 31-10). Although this and other experimental studies show a clear mechanism of action for optimizing the interaction between AV and VV timing, confusion surrounds this area, and a clinical advantage of patient-specific timing optimization has not been convincingly demonstrated in clinical trials80–82 (see later).
Reverse Volumetric Left Ventricular Remodeling
Mechanical resynchronization reduces mechanical load heterogeneity throughout the left ventricle. Sustained improvement in ventricular mechanics results in regression of adverse LV remodeling, termed reverse remodeling. This is indicated by reductions in LV volumes and mass, reduced mitral orifice size, regression of asymmetric hypertrophy, and increased EF.73,83–90 The magnitude of the reduction in end-systolic volume ranges between 10% and 30%,83,87,91,92 which is equivalent or greater than the effects of beta-adrenergic blockers and angiotensin-converting enzyme (ACE) inhibitors. These hemodynamic effects are chronically sustained assuming LV pacing is continuously maintained.73 Although improvement in New York Heart Association (NYHA) functional class, quality of life, and 6-minute walk tests may be confounded by placebo effects and reporting bias, reverse remodeling is a completely objective measure. More importantly, it appears to be related to better survival.92,93
Evidence of greater baseline electromechanical asynchrony and acute mechanical resynchronization appears to be necessary for reverse remodeling to occur, and the greater the reduction in asynchrony, the higher the probability of remodeling.72,90,94 Likewise, absence of electromechanical resynchronization acutely eliminates the possibility of chronic reverse remodeling.72,90 In canine hearts with induced LBBB, 8 weeks of CRT was sufficient to reverse almost completely the about 25% LV cavity dilation and asymmetric LV hypertrophy induced by dyssynchrony to pre-LBBB baseline values.42 This observation has been replicated in clinical studies where in some cases, reverse remodeling results in complete normalization of LV volumes and EF.64 This suggests that ventricular conduction delay may be the primary cause of DCM in some patients,63–65 as it was to a more moderate degree in the LBBB dogs.
Reduction in Functional Mitral Regurgitation
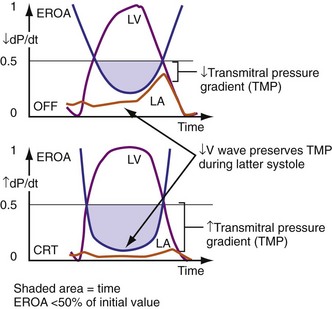
Figure 31-13 One mechanism for acute reduction in severity of functional mitral regurgitation during CRT.
(Modified from Breithardt OA, et al: Acute effects of cardiac resynchronization therapy on functional mitral regurgitation in advanced systolic heart failure. J Am Coll Cardiol 203:765-770, 2003.)
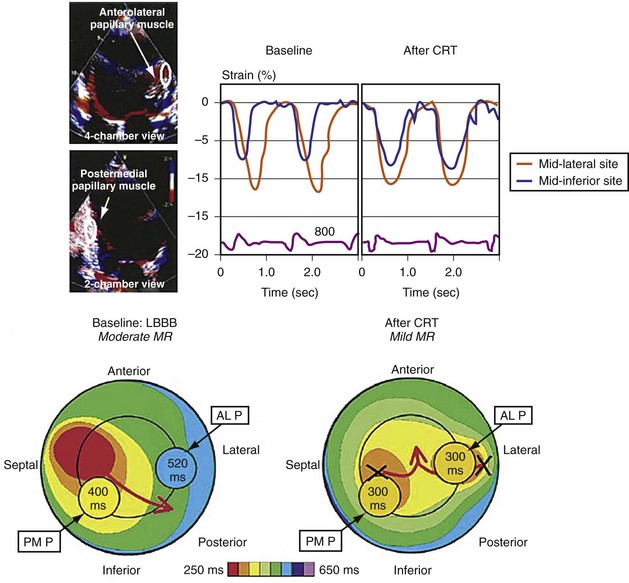
Figure 31-14 Effect of earlier delivery of posterior apparatus on functional mitral regurgitation (fMR).
(Modified from Kanzaki H, et al: A mechanism for immediate reduction in mitral regurgitation after cardiac resynchronization therapy: insights from mechanical activation strain mapping. J Am Coll Cardiol 44:1619-1625, 2004.)
 Cardiac Resynchronization Therapy Systems
Cardiac Resynchronization Therapy Systems
Hardware Systems
Leads and Electrodes: Non–Independently Programmable Ventricular Polarity Configurations
In a dual-bipolar configuration, both lead tips are the active electrodes (cathodes) and the rings are the common (nonstimulating) anode. However, the type of ventricular leads implanted defines the pacing/sensing vector (Figs. 31-15 and 31-16). With two unipolar leads, the bipolar setting results in no pacing or sensing. If both leads are bipolar, both rings act as the common electrode. If one lead is bipolar (RV) and the other lead is unipolar (typically LV), the ring on the bipolar lead acts as the common electrode (nonstimulating anode). This configuration results in shared-ring bipolar pacing and sensing. This hybrid bipolar/unipolar stimulation configuration (“dual cathodal”) is employed in most contemporary CRT pacing systems.
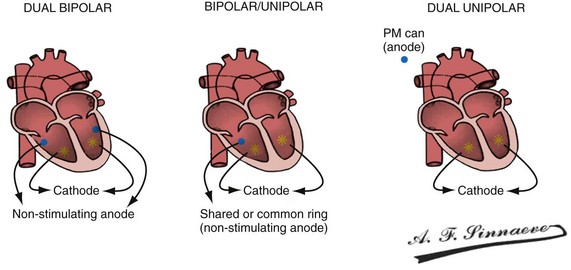
Figure 31-15 Biventricular pacing configurations.
(From Barold SS, Stroobandt RX, Sinnaeve AF: Cardiac pacemakers step by step: an illustrated guide. Malden, Mass, 2004, Blackwell.)
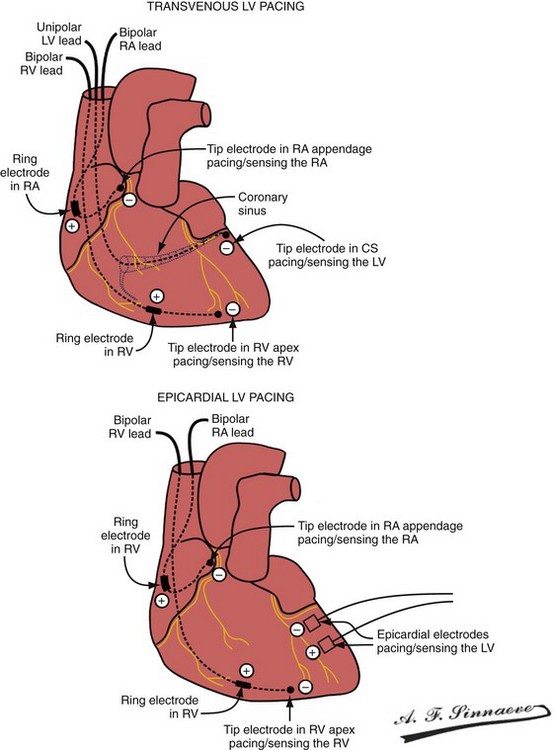
Figure 31-16 Leads and electrodes for biventricular pacing.
(From Barold SS, Stroobandt RX, Sinnaeve AF: Cardiac pacemakers step by step: an illustrated guide. Malden, Mass, 2004, Blackwell.)
Pulse Generators
Conventional dual-chamber pulse generators or specially designed multisite pacing pulse generators may be used for CRT applications (Fig. 31-17). A conventional dual-chamber pulse generator is well suited for CRT in patients with permanent atrial fibrillation. In this situation, the ventricular port is used for the RV lead and the atrial port is used for the LV lead. This permits programming of independent outputs and ventricular-ventricular timing by manipulation of the AV delay. The programming mode can be either DDD/R or DVI/R (see later). A conventional dual-chamber pulse generator can also be used for atrial-synchronous BiV pacing. The single ventricular output must be divided to provide simultaneous stimulation of the RV and LV (dual cathodal system with parallel outputs). This is achieved with a Y adapter and results in simultaneous RV and LV sensing, which may result in ventricular double-counting and loss of CRT, or pacemaker inhibition in the case of LV lead dislodgment into the coronary sinus with sensing of atrial activity.96
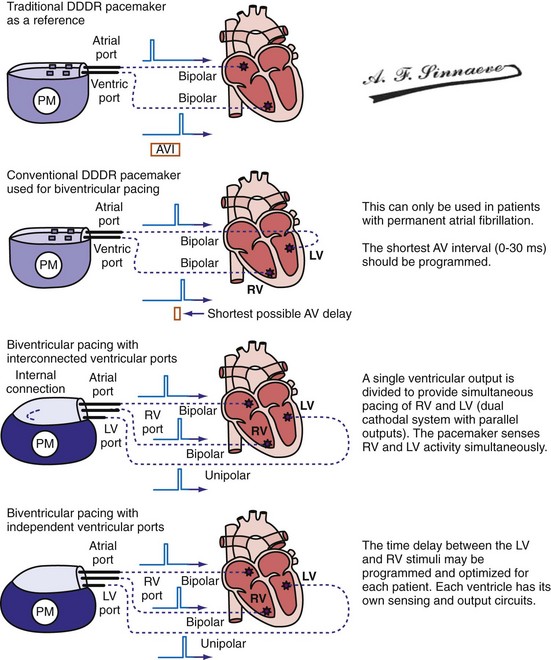
Figure 31-17 Pulse generators for biventricular pacing.
(From Barold SS, Stroobandt RX, Sinnaeve AF: Cardiac pacemakers step by step: an illustrated guide. Malden, Mass, 2004, Blackwell.)
Programming Considerations
Pacing Modes
It is axiomatic that for maximal delivery of CRT, ventricular pacing must be continuous.
Determining LV and RV Capture: Importance of Electrocardiography
Monochamber RV apical (RVA) pacing generates right (R) → left (L) activation in the frontal plane and anterior (A) → posterior (P) activation in the horizontal plane, resembling LBBB activation (Fig. 31-18). Consequently, mean QRS frontal plane axis is usually left superior and is infrequently right superior. Pacing from the midsuperior RV septum also generates right (R) → left (L) activation in the frontal plane and anterior (A) → posterior (P) activation in the horizontal plane, but the mean QRS frontal plane axis is typically left inferior. Monochamber LV pacing produces a variety of activation sequences depending on stimulation site and conduction blocks. A few generalizations are possible. Posterior wall stimulation usually generates an L → R, P → A activation, and mean QRS frontal plane axis is usually right superior, less often right inferior. A detailed discussion of ventricular activation sequences during monochamber LV and BiV pacing is provided later. Figure 31-19 shows a typical example of ventricular activation during monochamber RVA versus monochamber LV capture threshold determination.
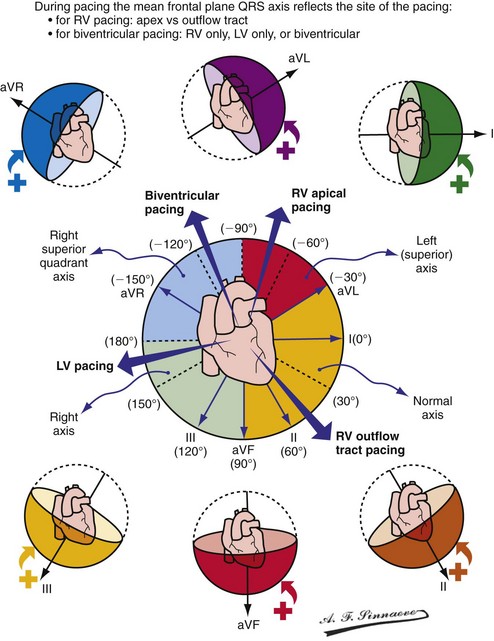
Figure 31-18 Mean frontal plane QRS axis during various conditions of ventricular pacing.
(From Barold SS, Stroobandt RX, Sinnaeve AF: Cardiac pacemakers step by step: an illustrated guide. Malden, Mass, 2004, Blackwell.)
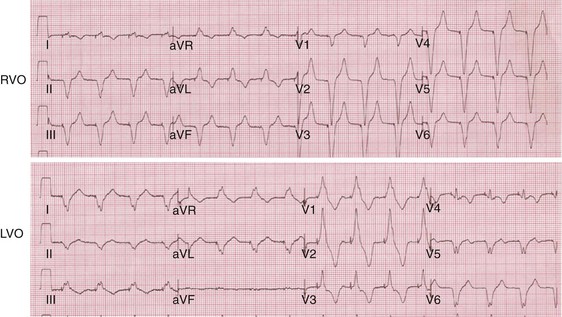
Figure 31-19 Typical ventricular activation sequences during monochamber RV and LV pacing threshold determination.
Anodal capture results in a distinct change in activation pattern compared to BiV pacing that can only be appreciated on the 12-lead ECG (Fig. 31-20). The change in QRS morphology related to loss of anodal capture as voltage output is decremented during a temporary threshold test using a single ECG lead may be misinterpreted as loss of LV capture and may result in erroneous overestimation of the LV threshold.
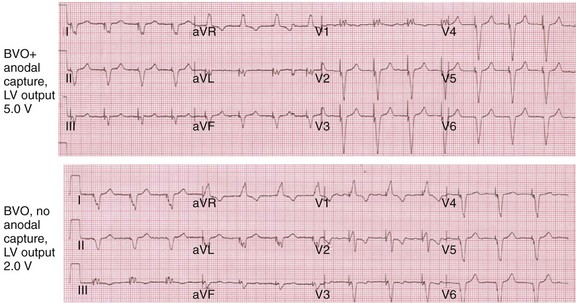
Figure 31-20 Effect of anodal capture on ventricular activation sequence during simultaneous biventricular (BiV) pacing.
Same patient as in Figure 31-19. RV apex and LV posterior wall capture thresholds are less than 1 V. Top, Simultaneous BiV pacing when LV output is 5 V (5 times > threshold) resulting in anodal capture. Bottom, Simultaneous BiV pacing when LV output is 2 V and no anodal capture. Note change in activation sequence. During high-LV-output BiV pacing, anodal capture is indicated by attenuation of monophasic R wave in V1 and loss of R (QS) in V2 (horizontal plane) and attenuation of QS in lead I and aVL (frontal plane). QRS duration is shorter. During lower-output BiV pacing, loss of anodal capture is indicated by dominant QS in I, aVL, and dominant R (V1) and RS (V2). QRS duration is longer.
The physiologic consequences of anodal capture are uncertain. One study demonstrated that anodal capture might be advantageous during CRT by counteracting the regional activation delay located at the inferior wall of the left ventricle and improving regional measures of intraventricular dyssynchrony.97
Automatic Adjustment of Left Ventricular Pacing Output
Operating Details
The LV (paced) → RV (sensed) conduction time is determined during cycles of up to eight (atrial synchronous) monoventricular pacing sequences (Fig. 31-21). To enhance accurate measurement of unidirectional ventricular conduction time, this test is initiated approximately 15 bpm above the ventricular rate measured during the rate and stability check. In dual-chamber modes, the pacemaker atrioventricular interval (pAVI) is shortened to 30 msec. These two adjustments are intended to guarantee complete replacement of AV and VV conduction with paced activation. Additionally, the blanking period on the RV sense amplifier is minimized to enhance RV sensing. LV-RV conduction is performed with LV output at the prevailing programmed setting. If RV sensing does not occur in response to a LV-only test stimulus, a backup BV pacing stimulus with LV at maximum amplitude occurs on the following cycle, and LV output remains at maximum amplitude except for single-test cycles.
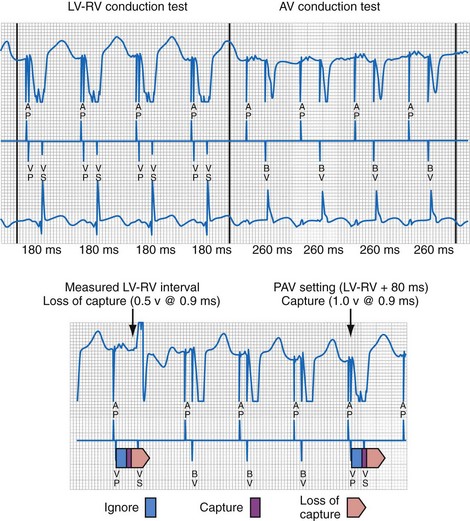
Figure 31-21 Left ventricular capture management (LVCM) operation.
(Modified from Crossley GH, et al: Automated left ventricular capture management. Pacing Clin Electrophysiol 30:1190-1200, 2007.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


