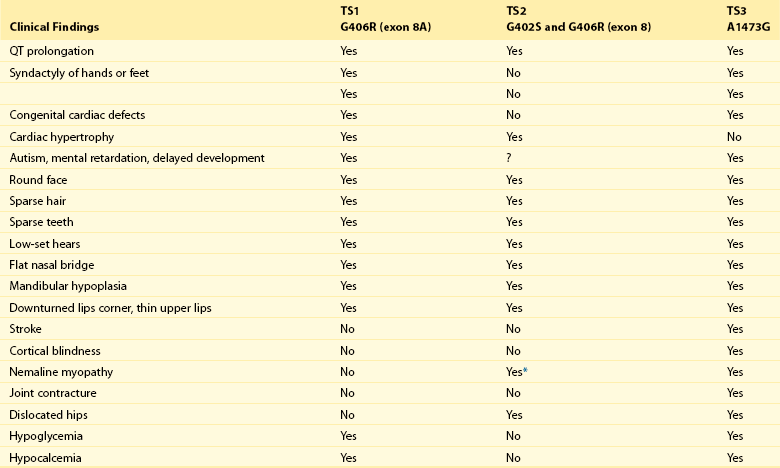
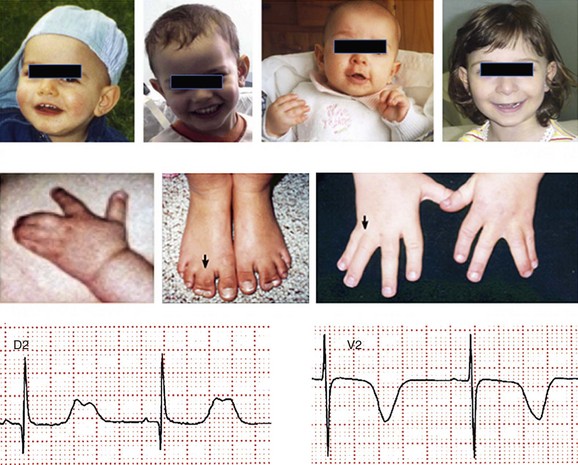
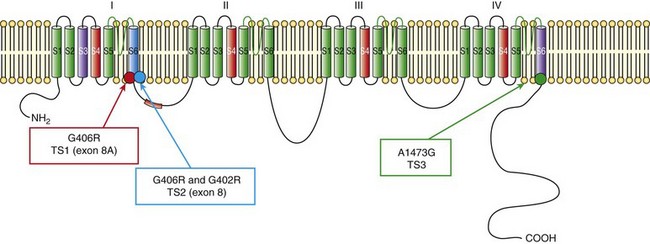
95 In 1992, Reichenbach et al.1 reported a case of intrauterine bradycardia followed by evidence of pronounced QT interval prolongation and a second-degree atrioventricular block after birth. The affected infant boy also had hand and foot syndactyly. He died suddenly at 5 months of age, and the investigators concluded that the phenotype could represent a novel clinical entity; this might have been the first case of TS identified in the literature. Few additional cases were reported thereafter,2 and all shared a severe phenotype, high mortality rate, and lack of familial recurrence. The reported cases and a larger cohort of newly identified patients were further analyzed to better define the spectrum of clinical manifestations. This clinical condition that was named Timothy syndrome to acknowledge the work of Katherine Timothy, a University of Utah and Harvard Medical School clinical coordinator, who for 15 years actively identified patients with the disease, helped to define new phenotypic features, and provided children and their families with clinical support and guidance.3 Fetal bradycardia is often the harbinger of the more severe clinical manifestation of the syndrome that often develops shortly after birth. The syndrome is characterized by a markedly prolonged ventricular repolarization (QT interval), 2 : 1 functional atrioventricular conduction block, and, in the majority of the patients, syndactyly of hands or feet, or both. The latter feature most often facilitates the diagnosis of the syndrome (Figure 95-1). Nonetheless, it is important to consider that the combination of syndactyly and QT prolongation can also occur in another inherited arrhythmogenic disease, Andersen-Tawil syndrome.4 This disorder, also defined as LQT7, is characterized by QT prolongation, ventricular arrhythmia, periodic paralysis, and facial dysmorphisms. Tristani-Firouzi et al.5 reported that syndactyly is present in approximately 10% of patients with Andersen-Tawil syndrome. Figure 95-1 Clinical presentation of Timothy syndrome: multisystem anomalies and developmental defects. The dysmorphic facial features (upper panel) typically include round face, spaced teeth, flat nasal bridge, receding upper jaw, and thin upper lip. Webbing of the toes and fingers (syndactyly) is observed in the majority of patients (middle panel). The electrocardiogram in the lower panel shows severe QT interval prolongation and giant T waves. The overall clinical presentation of TS is complex and encompasses several cardiac and extracardiac abnormalities (Table 95-1, see Figure 95-1). Despite the limited number of patients, it seems that important genotype and phenotype correlations might exist in the disease as shown in Table 95-1. Repolarization is remarkably prolonged in most patients, and the QTc often exceeds 550 ms.3 Besides the abnormal duration of repolarization, the morphology of the T wave is characterized by a set of abnormalities ranging from a prolonged ST segment followed by a small T wave (i.e., a pattern similar to that reported as typical for the LQT3 patients) to giant negative T waves (see Figure 95-1) and macroscopic T wave alternans. Congenital cardiac defects are present in approximately 60% of the patients; cardiac hypertrophy and ventricular dilatation are reported in more than 50% of patients.3,6,7 A mortality rate of 58%, with a mean age at death of 2.5 years, has been reported.3 In the early reported series of patients with TS, there were no adult individuals; therefore, the concept that the disease had a high lethality in first years of life became widespread. Currently, however, some adult cases of the disease have been reported.8 For the last 15 years, the authors have been following a female patient with TS and a mild cardiac phenotype who has reached the age of 32 years without manifesting cardiac arrhythmias. She has a reasonable social life and a QTc of 510 ms with syndactyly, but no structural cardiac abnormalities. In 2006, data from a population of 21 patients with TS9 showed that ventricular tachyarrhythmias, including ventricular tachycardia and ventricular fibrillation, occurred in approximately 80% of patients and constituted the most frequent cause of death. Functional atrioventricular block leading to bradycardia has been documented in 85% of patients. However, bradyarrhythmias have not played a role in cardiac-related deaths in TS. No specific trigger for cardiac arrhythmias has been reported, although recent experimental evidence (discussed later) suggests a role for adrenergic activation. Anecdotal reports have described the occurrence of life-threatening arrhythmias during anesthesia, but no information is available on specific anesthetics that might be dangerous to these patients.10 Non–arrhythmia-related deaths can also occur in TS. Among the 21 patients followed at the Molecular Cardiology Department of the Maugeri Foundation in Pavia, Italy, one died of sepsis and two died of malignant hypoglycemia that did not respond to glucose infusion in the hospital.3,9 The ST-T wave morphology in patients with TS can resemble that in patients with long QT syndrome with sodium channel mutations (the LQT3 variant, see Chapter 93). Based on the similarity in QT interval morphology between LQT3 and TS patients, it was hypothesized that the involvement of an increase in an inward current active during the plateau phase of the cardiac action potential. The hypothesis was confirmed when mutations in the gene encoding the α-subunit of the voltage-dependent L-type cardiac calcium channel (CACNA1C) on chromosome 12 were identified in SCN5A negative TS patients (Figure 95-2). Figure 95-2 Schematic diagram of the predicted transmembrane topology of the calcium channel, showing the location of mutations in the three variants of Timothy syndrome. The same G1216A transition in exon 8A (an alternatively spliced exon), which causes G406R amino acid substitution in transmembrane segment 6 of domain I of the protein (see later on), was identified in all patients with TS.3 In 2005, Splawski et al.3 reported two additional mutations in exon 8 (TS2): one of the two mutations is the same position G406R that was initially discovered in exon 8a. The child carrier of the G406R mutation in exon 8 did not have syndactyly, suggesting that the same mutation located in alternatively spliced exons could lead to different clinical manifestations. The second novel TS mutation identified by Splawski et al.,3 G402S, was located in an adjacent region of exon 8 and was identified in a child with a multifaceted disease without syndactyly. The individual with G402S was a mosaic and exhibited a milder phenotype than the girl with the G406R. Recently different mutation, A1473G (TS3), located in the transmembrane segment 6 of domain IV was identified by Gillis et al.11 This genetic variation is located in the terminal portion of transmembrane segment 6 of domain IV—that is, in a position similar to the other three mutations that are located in the sixth transmembrane segment of domain I (see Figure 95-2
Timothy Syndrome
Historical Notes
Phenotype and Natural History
Cardiac Events and Mortalityin Timothy Syndrome
Genetics of Timothy Syndrome
Mutations in the CACNA1C Gene
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Timothy Syndrome