Chapter 8
Thrombosis in Diabetes and Its Clinical Management
Introduction
The development of occlusive thrombotic vascular disease has become one of the major causes of morbidity and mortality in the modern world. Subjects with both type 1 and type 2 diabetes are at increased risk of developing cardiovascular disease, with approximately three-quarters of patients with diabetes ultimately dying from vascular causes. In the arterial system, subjects with diabetes have an increased prevalence of stroke, acute coronary syndromes, and peripheral vascular disease, while in the venous system a small increase in venous thrombotic disease has been observed, much of which may be related to associated comorbidities. Arterial disease is a chronic process characterized by the early development of endothelial dysfunction and fatty streaks followed by plaque formation, plaque instability, and occlusive thrombus formation on a ruptured plaque. Diabetes can affect all aspects of these processes, and clinical studies indicate that coronary artery plaques from subjects with diabetes have increased plaque thrombus and monocyte/macrophage infiltration compared to nondiabetes controls [1]. This, together with more extensive disease affecting both the proximal and distal coronary vasculature, describes a situation in which the circulation supplying the heart has more lesions, with a greater propensity to rupture and to produce more thrombus. The arterial clot is characterized by the development of a platelet-rich fibrin mesh, the fibrin being generated by activation of the fluid phase of coagulation, while venous thrombosis is characterized by a fibrin-rich, platelet-poor thrombus. Type 2 diabetes is associated with increased platelet activation [2], and with a range of abnormalities in coagulation and fibrinolysis related to the metabolic abnormalities associated with insulin resistance and hyperglycemia [3]. These prothrombotic changes contribute to the increased prevalence of acute coronary syndromes and other arterial disorders; increased platelet reactivity in particular has been reported to prospectively predict risk of major adverse cardiovascular events in type 2 diabetes patients with stable coronary artery disease [4]. Most evidence seems to indicate that thrombotic disorders start to appear with the development of insulin resistance and in the presence of advanced complications such as chronic kidney disease. Glycemia has an additional effect on many of these processes, which tend to deteriorate as the chronic nature of diabetes unfolds. Clinical studies suggest that as a consequence, uncomplicated type 1 diabetes has relatively minor alterations in thrombotic profile, while nondiabetes insulin-resistant relatives of subjects with diabetes have clustering of inflammatory thrombotic risk prior to the appearance of frank hyperglycemia [5–7]; and in both groups further changes occur as the disorder progresses.
The recognition that myocardial infarction usually results from thrombus formation on a ruptured plaque led to a revolution in therapeutic approaches that has improved primary and secondary prevention of cardiovascular disease as well as the management of acute coronary syndromes. Among these, the development of increasingly sophisticated inhibitors of platelet activation, direct thrombin inhibitors, and heparin-like molecules have transformed care of both diabetes and nondiabetes subjects with coronary artery disease. In this chapter we will describe the mechanisms that underpin abnormalities in platelet function and the fluid phases of coagulation and fibrinolysis in subjects with diabetes, the way in which these changes relate to cardiovascular disease, and how antiplatelet agents and anticoagulants ameliorate cardiovascular outcomes in subjects with diabetes.
Mechanisms of Thrombosis
The hemostatic system consists of a fluid phase of activators and inhibitors of coagulation and fibrinolysis that regulate the formation and breakdown of fibrin and a cellular, platelet phase that interacts with sites of vascular damage and fibrin to release a range of procoagulant and inflammatory mediators. Thrombin is the pivotal enzyme in the coagulation pathway, having a crucial role in both fibrin formation and platelet activation. Thrombin is generated by the cleavage of prothrombin by a Factor Xase complex, which occurs as the result of interactions between tissue factor-activated Factor VII and Factor X secondary to vascular damage. Thrombin, while having major procoagulant and pro-inflammatory effects, can express an anticoagulant effect when thrombin binds to the cell-associated receptor thrombomodulin to change thrombin’s substrate [8].
Fibrinogen Cleavage
Fibrinogen is a large protein produced by the liver that consists of two sets of three α, β, and γ chains linked by disulfide bonds [9]. Thrombin cleaves fibrinogen by cutting small fibrinopeptides from each of the fibrinogen α chains, allowing the α chains to open out and interact with other cleaved fibrinogen molecules, leading to the formation of double-stranded fibrils that branch out to create a complex fibrin network. Cleavage of fibrinopeptide B allows lateral aggregation of the developing fibrin structure.
Factor XIII Activation and Fibrin Cross-linking
Coagulation FXIII is a transglutaminase that circulates in plasma in a heterodimeric structure consisting of two A and two B subunits. Thrombin activates Factor XIII by cleaving a 37 amino acid peptide from the A subunit, which promotes separation of the A and B subunits and permits exposure of the active site on FXIIIA. Activated Factor XIIIA covalently cross-links fibrin fibrils, which creates a fibrin structure that is insoluble, with altered mechanical properties and increased resistance to fibrinolytic activity [10].
Fibrinolysis
Analogous to thrombin, plasmin is the pivotal enzyme in the fibrinolytic cascade. Plasmin is generated by the cleavage of plasminogen by tissue plasminogen activator (tPA), and this reaction occurs 1,000-fold faster in the presence of fibrin. A lysine binding site on plasmin binds plasmin to fibrin, which facilitates fibrin breakdown and also protects plasmin from local inhibition by antiplasmin. Plasmin cleaves arginine and lysine sites on a range of molecules and its activity is tightly controlled by antiplasmin to prevent systemic proteolysis [11]. Cleavage of fibrin by plasmin leads to the generation of fibrin degradation products, which can be measured in plasma, and one of which, D-dimer, is used as an indicator of the presence of venous thrombotic disease. In addition to antiplasmin, other inhibitors of this pathway include plasminogen activator inhibitor-1 (PAI-1) and thrombin activatable fibrinolysis inhibitor (TAFI). PAI-1 is the fast-acting inhibitor of tPA that binds to and inhibits tPA activity. PAI-1 is produced by endothelial cells and platelets and circulates in plasma in excess over tPA, and is also found in fairly high concentrations in thrombus. TAFI is found in large quantities in platelets and plasma, and is activated by thrombin, a cleavage event that is much enhanced when thrombin is bound to thrombomodulin. Activated TAFI cleaves the N-terminal lysine residues from degrading fibrin to prevent binding of plasminogen and tPA to fibrin, which results in inhibition of plasmin generation and clot lysis [12].
Platelet Activation
Damage to the vascular wall leads to two key events in platelet associated clot formation: 1) receptor-mediated platelet adherence and aggregation; and 2) thrombin-mediated platelet activation. Adherence to the subendothelial matrix is facilitated by a range of glycoprotein receptors (GP Ib/IX, GPVI, and GPIa), which interact with von Willebrand factor to promote platelet adhesion. This interaction leads to activation of platelet GPIIb/IIIa, which binds fibrinogen and promotes platelet aggregation. Thrombin is the most potent platelet activator, which exerts its effects through binding to protease-activated receptor 1 (PAR-1) on the platelet surface. This leads to a cascade of signaling processes, culminating in the release of a range of inflammatory and thrombotic mediators, which further promote clot formation. In addition to thrombin, a range of other mediators, including ADP, collagen, and thromboxane, can activate the platelet through a receptor-binding event. These receptors provide some of the novel targets for therapeutic approaches described later and are discussed in a number of excellent reviews [13–15]. The main steps in clot formation and lysis are summarized in Figure 8.1.
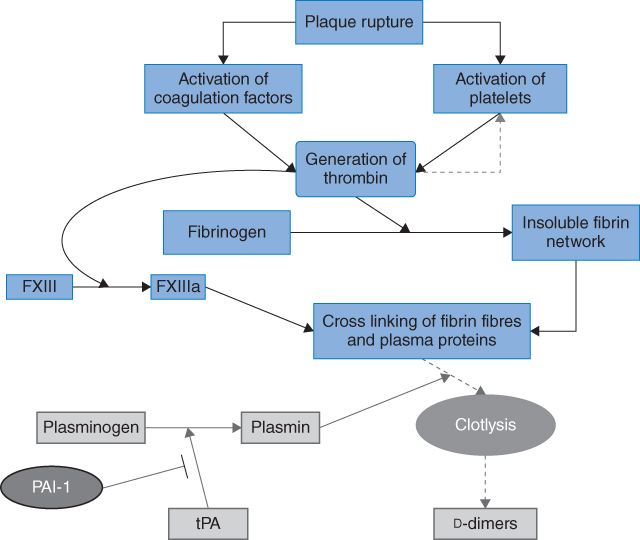
Figure 8.1 Clot formation and fibrinolysis. Rupture of an atherosclerotic plaque exposes a prothrombotic core, resulting in activation of platelets and coagulation proteins. Thrombin is formed with subsequent conversion of soluble fibrinogen to insoluble fibrin, which is further strengthened by thrombin-activated FXIII. Thrombin further activates platelets, enhancing the thrombotic process. Tissue plasminogen activator mediates conversion of plasminogen to plasmin, which lyzes the clot, generating d-dimers. Fibrinolysis is inhibited by a number of proteins, including plasminogen activator inhibitor (PAI)-1.
Summary of the Mechanisms of Thrombosis
In describing the individual components of these processes, it is easy to lose sight of the exquisite control that is exerted at all levels of clot formation. In addition to platelets, thrombosis involves binding events on endothelium, subendothelial layers, macrophages, and leukocytes, with the balance between thrombosis and clot lysis and the localization of clot formation depending on these interactions. Emerging evidence demonstrates the importance of thrombotic inflammatory interactions, both at the cellular level where platelet/macrophage binding initiates the release of a range of soluble procoagulant and inflammatory molecules, and in the fluid phase where, for example, complement C3 binds fibrin to inhibit fibrinolysis [16]. As these events cycle toward fibrin formation and fibrin/platelet interactions, further levels of control are exerted by the interaction of activators and inhibitors of lysis on fibrin itself. All of these levels of control direct and limit thrombus formation and fibrinolysis to the site of need to prevent systemic thrombus formation and proteolysis.
Mechanisms of Thrombosis in Diabetes
Coagulation and Fibrinolysis
The major consistent hemostatic abnormality observed in insulin-resistant type 2 diabetes is marked suppression of fibrinolysis associated with increased levels of both PAI-1 and tPA [17]. Studies of euglycemic first-degree relatives of subjects with type 2 diabetes indicate that such individuals tend to be insulin resistant and have raised triglyceride, tPA, and PAI-1 before a diagnosis of diabetes is made; there is little evidence to suggest that glycemia influences this pattern. Several studies have reported strong associations between insulin resistance, triglyceride, and PAI-1 levels, and it appears that the severity of suppression of fibrinolysis clusters with an increasing number of conventional risk factors. The PAI-1 gene has a 4G/5G polymorphism 675 bp from the start site, and the 4G allele has been associated with both higher PAI-1 levels and increased risk of acute coronary syndrome [18]. Additionally, there are indications that interactions between the 4G/5G genotype and features of the metabolic syndrome regulate circulating PAI-1 levels, providing a path for increasing cardiovascular risk [19]. TAFI levels seem to be unaffected by insulin resistance or hyperglycemia, although there are indications that levels are increased in type 2 diabetes with microalbuminuria [20]. Coagulation Factor VII levels show a similar association with insulin resistance and type 2 diabetes is also associated with elevated fibrinogen and Factor XII levels [17]. The importance of insulin resistance in these early manifestations of thrombotic risk is emphasized by studies of insulin-sensitizing agents, which consistently demonstrate that metformin and thiazolidinedione use is associated with reductions in PAI-1 and tPA, while metformin has additionally been reported to lower levels of Factors VII and XIIIA [17].
Clot Structure
Jörneskog reported changes in clot structure in type 1 diabetes subjects showing reduced permeability to indicate a more compact structure [21]. Other studies have made similar observations using plasma-purified fibrinogen from type 2 diabetes patients [22]. The evidence indicates that posttranslational modifications to fibrin(ogen) are promoting structural alterations to fibrin [23]. However, such changes facilitate decreased plasmin generation on the clot surface and increased antiplasmin binding, with an overall effect on fibrinolysis rather than clot formation [24]. Compact structures have been associated with increased cardiovascular risk and poorer cardiovascular outcome in nondiabetes populations, and it is likely that a range of metabolic influences affect this phenotype.
Platelet Activation
The circulating platelet is sensitive to a wide range of metabolic changes associated with diabetes, of which hyperglycemia is the most clinically apparent. Short-term exposure to hyperglycemia increases platelet reactivity and improvements in glycemic control ameliorate these effects. It has been proposed that hyperglycemia may have osmotic effects on the platelet, alter protein kinase C expression, and/or have indirect effects through exposure to glycated proteins. In this respect, recent evidence indicates that AGE proteins induce a prothrombotic state through interactions with the platelet CD36 receptor mediated by a JNK2 pathway [25]. Oxidized LDL is reported to activate platelets in insulin-resistant subjects [26], and CD36 is involved in platelet activation through interactions with dyslipidemia and oxidative stress, effects that are absent in CD36 null mice [27]. It is interesting to note that the macrophage CD36 receptor is well established as having a role in the formation of early fatty streaks through interactions with oxidized LDL leading to increased foam cell formation. This response in macrophages is accentuated in insulin-resistant states and is ameliorated by thiazolidinediones. Thiazolidinediones are reported to possess antiplatelet effects, although it is not known whether this effect on platelets is mediated to any extent through effects on CD36. Other potential influences include effects of insulin resistance. Insulin has anti-aggregatory effects in platelets from insulin-sensitive subjects and emerging data indicate that IGF-1 may have prothrombotic effects on the platelet through interaction with the hetrodimerized insulin/IGF-1 receptor in insulin-resistant states. In a population of 208 type 2 diabetes patients with stable coronary artery disease followed up for 24 months, carriers of a particular insulin receptor substrate-1 (IRS-1) genotype exhibited both increased platelet reactivity and a significantly higher risk of major adverse cardiovascular events [28]. These findings both implicate the insulin-signaling pathway in cardiovascular outcomes and provide a potential mechanism for inter-individual differences between subjects with diabetes.
Overall, the available data indicate that diabetes is associated with a range of metabolic abnormalities that adversely influence platelet function. Management of the platelet aspect of this prothrombotic state should involve normalization of the metabolic changes seen in diabetes and the appropriate use of antiplatelet therapy, as discussed below.
Management and Prevention of Thrombotic Events in Diabetes
Individuals with diabetes are at increased risk of cardiovascular events and their prognosis following vascular ischemia is worse than the nondiabetes population. This increase in mortality is related to a combination of more extensive vascular pathology associated with increased thrombotic milieu, secondary to enhanced platelet activation and quantitative/qualitative changes in procoagulant and antifibrinolytic factors. The detailed discussion of the management of vascular ischemic events is covered elsewhere in this book; we will concentrate on highlighting diabetes-specific antithrombotic therapy.
Antiplatelet Agents
There are a number of antiplatelet agents in use for the treatment and prevention of cardiovascular disease in diabetes, which mainly affect three pathways of platelet activation, although agents are under development that target additional pathways. In this section the various antiplatelet agents used in the treatment and prevention of cardiovascular disease are discussed, with an emphasis on the role of these agents in diabetes.
Aspirin
Aspirin acetylates serine residue 529 in cyclo-oxygenase (COX)-1, irreversibly inhibiting enzyme activity and blocking the production of thromboxane A2, resulting in diminished platelet aggregation. Another mode of action that we and others have shown is that aspirin acetylates fibrinogen, altering fibrin network characteristics, making the clot easier to lyze [29–32]. Also, aspirin may influence clot lysis indirectly through a nitric oxide-dependent mechanism [33, 34]. These platelet-independent fibrinolytic properties of aspirin are potentially important clinically, and may explain the enhanced fibrinolytic effects of streptokinase when used with aspirin in the ISIS-2 study [35].
Aspirin is regularly used in the setting of acute coronary syndrome (ACS), the benefit of which has been repeatedly demonstrated in individuals with or without diabetes [35–37]. Aspirin should be given as early as possible in ACS, regardless of whether the diagnosis is unstable angina/NSTEMI (non-ST-elevation myocardial infarction) or STEMI (ST-elevation myocardial infarction), at an initial dose of 162–325 mg (300 mg is used in the UK) with a combination of other anti-thrombotic agents (detailed below). This is followed by a maintenance dose of 75–162 mg/day (75 mg/day in the UK) in those with proven vascular pathology. In the longer term, aspirin is used for secondary cardiovascular protection in diabetes [38, 39], a practice supported by two large meta-analyses [40, 41]. The percentage reduction in vascular ischemia with the use of aspirin for secondary prevention is 17% (from 22.3 to 18.5%; p < 0.01) and 22% (from 16.4 to 12.8%; p < 0.00001) in subjects with and without diabetes respectively. These data indicate that aspirin may be less effective in secondary cardiovascular protection in diabetes, a concept supported by a relatively recent observational study failing to show a benefit for aspirin in secondary prevention [42].
The use of aspirin for primary cardiovascular protection in diabetes is more controversial. Until recently, aspirin has been regularly used in these circumstances, although the evidence supporting such practice was surprisingly scarce. Indeed, several pieces of evidence suggest that aspirin should not be used in all diabetes subjects for primary prevention. In the Primary Prevention Project (PPP) trial, aspirin treatment failed to offer significant cardiovascular protection in diabetes patients, in contrast to individuals with no diabetes [43]. A meta-analysis of more than 140,000 subjects has shown that the use of antiplatelet agents (mainly aspirin) resulted in a 22% reduction in cardiovascular events, but in a subgroup of around 5,000 diabetic subjects the risk reduction was only 7%, which was not statistically significant [41]. Moreover, two recent primary prevention studies, JPAD and POPADAD, failed to show an impact of aspirin on cardiovascular events in individuals with diabetes [44, 45]. However, JPAD demonstrated an overall benefit in the older population, suggesting that a subgroup of patients may benefit from this therapy. A longitudinal observational study of 651 diabetes individuals over 11.6 years’ follow-up period (7,537 patient-years) has shown a reduction in CV events in aspirin-treated subjects after adjustment for significant CV variables (HR 0.30 CI 0.09–0.95), indicating that aspirin may be beneficial in some patients with diabetes [46]. In contrast, an increase in cardiovascular events was reported in aspirin-treated Chinese diabetes subjects with no history of ischemic heart disease [47]. Similar results were documented in the Swedish record linkage study, although again, a beneficial effect was observed in the older population [48]. A meta-analysis of seven studies, including 11,618 diabetes individuals, reported a 9% reduction in overall major adverse cardiovascular events (MACE) without an effect on mortality, which may be due to the relatively short period of follow-up [49].
Data indicate that the efficacy of aspirin in primary prevention in diabetes is compromised and should not be used in all patients. However, it appears that some individuals with diabetes, perhaps those at high cardiovascular risk, benefit from aspirin therapy for primary prevention. Given this situation, current national and international guidelines limit the use of aspirin for primary prevention in diabetes to individuals at “high cardiovascular risk” without clearly categorizing this group and leaving the decision at the discretion of the attending physician. There are two ongoing outcome studies investigating the appropriate use of aspirin for primary cardiovascular protection in diabetes (ASCEND and ACCEPT-D, clinical trial registration number NCT00135226 and IS-RCTN48110081 respectively), which are expected to report in the next three to four years.
In summary, 1) aspirin continues to be used in ACS (inclusive of unstable angina, NSTEMI, and STEMI), in combination with other antiplatelet agents in both diabetes and nondiabetes subjects; 2) aspirin is used as monotherapy for secondary cardiovascular protection, but may be less efficacious in subjects with diabetes; 3) there is no convincing evidence for the use of aspirin monotherapy for primary cardiovascular protection in diabetes, although some guidelines recommend its use in high-risk subjects.
Clopidogrel
Clopidogrel, a thienopyridine agent, is an irreversible antagonist of the platelet P2Y12 receptor. Clopidogrel is a pro-drug and is converted to the active metabolite by the P450 system in the liver; the onset of action may be delayed by CYP genetic polymorphisms. Clopidogrel is used in combination with aspirin in subjects with acute coronary syndrome and as monotherapy in those intolerant to aspirin or in patients with symptomatic cerebrovascular disease despite aspirin therapy [50, 51].
The combination of aspirin and clopidogrel in the setting of ACS (usually 300–600 mg loading dose of clopidogrel followed by maintenance of 75 mg/day) has been established through a number of large-scale clinical trials, with benefits shown in diabetes and nondiabetes subjects [52–55]. However, newer agents have recently shown superior efficacy (detailed below), and dual therapy with clopidogrel is now used less frequently in some centers, where the cost of the newer agents can be absorbed. Combination aspirin and clopidogrel is usually given for a year following ACS (clopidogrel at 75 mg/day) and aspirin monotherapy continued beyond this period. However, routine combination therapy in high-risk diabetes individuals with no recent ACS is not recommended due to the absence of clinical benefit, supported by data from the CHARISMA trial [56].
There is evidence to suggest that clopidogrel monotherapy is superior to aspirin when used for secondary cardiovascular prevention in diabetes. The CAPRIE study enrolled 19,185 subjects with established cardiovascular disease and randomized to aspirin 325 mg/day or clopidogrel 75 mg/day. In the post hoc analysis of the diabetes group, a 12% reduction in vascular ischemia was documented comparing aspirin with clopidogrel users (from 17.7% to 15.6%, respectively; p = 0.04); the difference was even more pronounced in insulin users [57]. However, as this was an unspecified post hoc analysis, it has largely gone unnoticed and the data failed to find their way into clinical practice.
It should be noted that variability in the biochemical efficacy of clopidogrel is largely due to variability in drug metabolism. The prevalence of clopidogrel low responsiveness varies widely between studies (5–40%), related to the test used, definition of resistance, and population studied [58]. Diabetes is one factor accounting for the reduced response to clopidogrel, particularly in the presence of poor diabetes control, microvascular complications, or in those having insulin treatment [59]. Some of the suggested mechanisms for the reduced efficacy of clopidogrel in diabetes include insulin resistance resulting in diminished platelet response to insulin, upregulation of P2Y12 receptor signaling, and higher platelet turnover.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


