Chapter 10 Thrombosis
Overview of Thrombosis
When the endothelial surface becomes damaged, however, release of many procoagulant proteins (especially tissue factor) and activation of platelets result in uncontrolled hemostasis at the site of vascular injury.1 As the thrombus begins to form, it recruits additional platelets to the area, leading to further platelet activation. Initially, tethering of platelets is dependent upon exposure of glycoprotein (Gp)Ib-V-IX in damaged collagen, which binds to von Willebrand factor (vWF), resulting in adhesion of platelets to the area of injury. Further recruitment of platelets is mediated through activation of the GPIIb-IIa platelet receptor, which undergoes a conformational change leading to increased affinity for fibrinogen. These events culminate with further platelet activation that results in release of many essential components for thrombus formation, including adenosine diphosphate (ADP), serotonin, and thromboxane A 2 (TxA2).
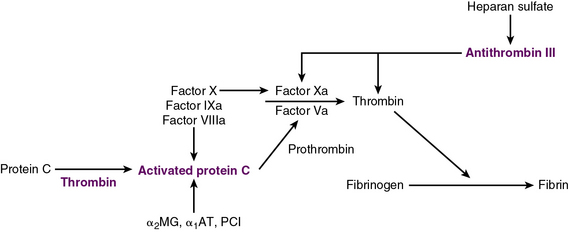
Exposure of vascular collagen also leads to activation of the normal mechanisms of hemostasis—including the coagulation cascade—through exposure of tissue factor, leading to “hemostasis in the wrong place.” The coagulation regulatory system is outlined in Figure 10-1 and discussed in detail in Chapter 5. Briefly, both the tissue factor–mediated pathway (extrinsic) and the contact-mediated pathway (instrinsic pathway) rely on activation of inactive enzyme precursors of serine proteases, which then reflexively lead to activation of another protein within the cascade. The ultimate step results in cross-linking of fibrin to stabilize a platelet plug, leading to thrombus formation. The tissue factor–initiated pathway is essential for thrombus formation. When tissue factor is released during cellular injury, factor VII is activated and complexes. This complex next activates factors X and XI. Activation of factor X is essential for conversion of prothrombin (factor II) to thrombin through the prothrombinase complex on activated platelets. This cascade of coagulation proteins is essential for hemostasis but also can have deleterious affects when it occurs unregulated, leading to unwanted thrombotic complications.
Platelets, Thrombosis, and Vascular Disease
Venous Thrombosis
Genetic risk factors associated with increased risk of VTE include mutations in factor V (Leiden) and prothrombin 20210, as well as mutations leading to deficiencies in antithrombin, protein C, and protein S. Approximately 5% of the Caucasian population has at least one mutation for factor V Leiden, and 15% to 20% of patients who present with a VTE carry the mutation.2–5 Approximately 2% of the population carry the prothrombin gene mutation, but it may be present in approximately 5% to 15% of persons with VTE.6 The population frequencies of mutations in other genes responsible for other coagulation factors (e.g., protein C) are estimated to be 1 in 500 individuals. Antithrombin III deficiency is associated with a frequency of 1 in 300 in the general population, and in 3% to 5% of those with thrombotic events. Previously it was thought that genetic mutations in genes important for methylene tetrahydrofolate reductase and hyperhomocysteinemia increased the risk of VTEs; however, recently this association has been shown to be less likely.7
One of the acquired risk factors known to be important in both venous and arterial thrombosis is acquisition of antiphospholipid antibodies, which represent a family of antibodies against phospholipids (e.g., cardiolipins) and phospholipid binding proteins (e.g., GpI β2). Mechanisms responsible for thrombosis are still speculative but may include inhibition of protein C, antithrombin, and annexin A5 expression; binding and activation of platelets; enhanced EC tissue factor expression; and activation of the complement cascade.8 Criteria for diagnosis of the associated disorder, antiphospholipid syndrome, includes the presence of both clinical events and laboratory evidence for the presence of antiphospholipid antibodies.9
Arterial Thrombosis
A primary mechanism of arterial thrombosis is rupture of atherosclerotic plaques, precipitating platelet-rich aggregates. Arterial thrombosis can have catastrophic consequences when it occurs in the coronary or carotid artery circulation. Factors that can exacerbate these types of thrombotic events include smoking, diabetes, hypertension, and hyperlipidemia. Thrombosis generally occurs when there is disruption in the hemostatic balance that results when pro- and anticoagulant molecules are at disequilibrium. Endothelial damage shifts this balance towards a more procoagulant force, leading to exposure of collagen and tissue factor. Collagen that is now exposed can activate platelets in the blood flowing through the vessel, and concomitantly thrombin is generated as the coagulation cascade is initiated in the presence of tissue factor. Genetic modifications of proteins important in coagulation can alter this process, creating a propensity to form thrombi in the arterial system (Box 10-1). These mutations affect platelet function, leading to increased propensity to aggregation.
 Box 10-1
Box 10-1


