Thermal Lung Injury and Acute Smoke Inhalation
INTRODUCTION AND EPIDEMIOLOGY
Smoke inhalation is a serious medical problem and continues to have a significant impact on the morbidity and mortality of patients with flame burns. According to the American Burn Association Repository (2012), inhalation injury is present in 17% of patients with flame burns and increases the overall mortality rate of these patients up to 24%, while the mortality of burn patients without inhalation injury is 3%.1 The presence of smoke inhalation injury prolongs the length of hospital stay 2.5-fold compared to those without smoke inhalation injury (24 days vs. 10 days).1
Similar percentages of fire victims who have sustained smoke inhalation appear in several other countries.2–5 In patients with combined injury, the lung is the critical organ and the progressive respiratory failure associated with pulmonary edema is a pivotal determinant of mortality.6–8 Although not as lethal, smoke inhalation alone is a serious problem. It is estimated by the World Health Organization that there are over one billion people who develop airway and pulmonary inflammation as a result of inhaling smoke from indoor cooking fires, forest fires, and burning of crops.9,10
The inhalation of smoke has been of interest for a number of years, especially as a result of the use of gas warfare. In the 1940s there were two very large fires that focused interest on the inhalation of smoke in fire victims. The first was a fire at a nightclub in Boston called the Cocoanut Grove, where a large number of people were trapped in a burning building and consequently sustained severe inhalation injury.11,12 It is interesting that in recent times a similar fire occurred in a nightclub near Boston in Rhode Island. The second occurred in Texas City across the bay from Galveston, Texas.13 Here a ship exploded in a harbor and set off a chain of explosions and fires among some 50 refineries and chemical plants, resulting in over 2,000 hospital admissions of patients with smoke inhalation alone, those with burn injuries, many of whom who had simultaneously inhaled smoke as well. At any rate these two disasters led to the establishments of centers for the care of burn victims and to research into the pathophysiology of burn injury. In many ways the burn victims of the 9/11 disaster were similar to these individuals since the burns and inhalation involved combustion of petroleum products.14,15 Approximately half (49%) of 790 victims who survived the World Trade Center attack had an inhalation injury.15,16
TOXIC SMOKE COMPOUNDS
The fire environment contains a number of toxic compounds, each of which is discussed in this section.
Inhalation injury is caused by steam or toxic inhalants such as fumes, gases, and mists. Fumes consist of small particles dispersed in air with various irritants or cytotoxic chemicals adherent to the particles. Mists consist of aerosolized irritant or cytotoxic liquids. Smoke consists of a combination of fumes, gases, mists, and hot air. Heat, toxic gases, and low oxygen levels are most common causes of death in a fire scene. A large variety of toxic gases and chemicals can be generated depending on the fire environment (Table 94-1).
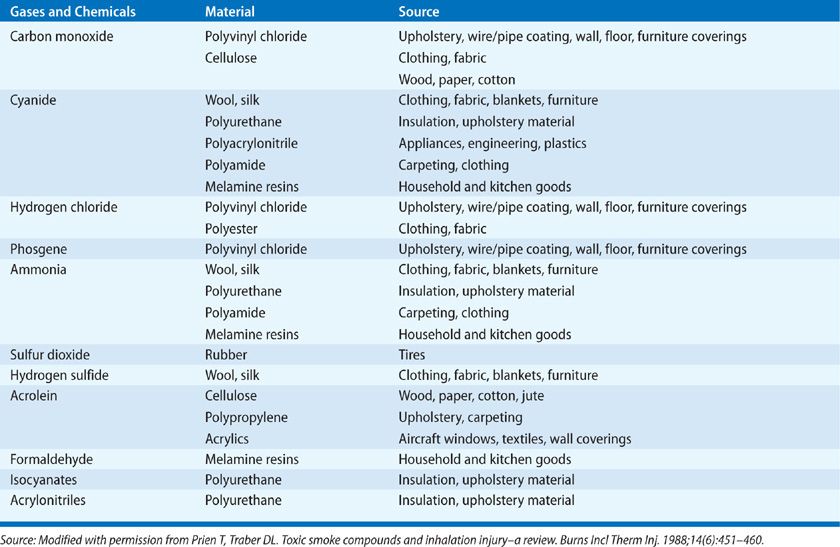
Many of these compounds may act together to increase mortality, especially carbon monoxide and hydrogen cyanide17,18 in which a synergism has been found to increase tissue hypoxia and acidosis18 and perhaps also decrease cerebral oxygen consumption and metabolism.19,20 Hydrogen sulfide would also be predicted to synergize with carbon monoxide since both cyanide and hydrogen sulfide are inhibitors of mitochondrial cytochrome oxidase. Victims may be incapacitated by the blinding and irritating effects of smoke, as well as the decreasing oxygen concentration that occurs with combustion and results in progressive hypoxia.
Inhalation injury can be classified as (1) upper airway injury, (2) lower airways and pulmonary parenchymal injury, and (3) systemic toxicity. The extent of inhalation damage depends on fire environment: the ignition source, temperature, concentration, and solubility of the toxic gases generated. For instance, thermal and chemical compounds usually cause upper airway injury. The water-soluble materials such as acrolein and the other aldehydes damage the proximal airways and set off reactions that are inflammatory to the bronchi and parenchyma, whereas agents with lower water solubility such as chlorine, phosgene, nitrogen oxide, and nitrogen dioxide or N2O3 or even N2O4 are more likely to cause insidious injury.21 Toxic gases such as carbon monoxide and cyanide rarely damage the airway but affect gas exchange, producing more systemic effects. Thus, it is important to obtain information relative to the source of the fire and the combustion products generated when treating a fire victim (see Table 94-1). It is also important to know the duration of exposure and the extent to which the fire victim was in an enclosed area because this relates to the dose of toxic materials presented.
 CARBON MONOXIDE
CARBON MONOXIDE
Carbon monoxide (CO) is an odorless, colorless gas that is produced by incomplete combustion of many fuels, especially cellulolytic (cellulose products) such as wood, paper, and cotton.22 Carbon monoxide toxicity remains one of the most frequent immediate causes of death following smoke-induced inhalation injury. There are 50,000 estimated annual visits to emergency department in the United States as a result of CO poisoning.23 The predominant toxic effect of CO is its binding to hemoglobin to form carboxyhemoglobin (COHb). The affinity of CO for hemoglobin is ~200 to 250 times higher than that of oxygen.24 Inhalation of a 0.1% CO mixture may result in generation of a COHb level as high as 50%. The correlation of inhaled CO and COHb levels is summarized in Table 94-2.
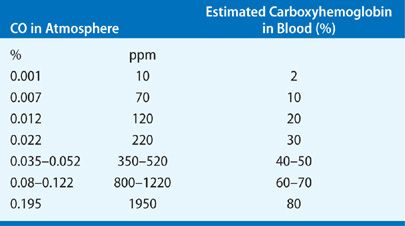
The competitive binding of CO to hemoglobin reduces delivery of oxygen to tissues leading to severe hypoxia, especially of most vulnerable organs such as brain and heart. The oxygen–hemoglobin dissociation curve loses its sigmoid shape and is shifted to the left, thus further impairing tissue oxygen availability.17 In addition, ability of CO to bind to intracellular cytochromes and to other metalloproteins contributes to CO toxicity. This competitive inhibition with cytochrome oxidase enzyme systems (most notably cytochromes a and P-450) results in an inability of cellular systems to use oxygen.27 Shimazu et al. have shown that extravascular binding of CO to cytochromes and other structures accounts for 10% to 15% of total-body CO stores, which explains the two-compartment elimination of CO from the circulation.28 CO also inhibits cytochrome c oxidase activity in lymphocytes. The electron chain dysfunction by CO may cause electron leakage, leading to superoxide production and mitochondrial oxidative stress. Additionally, the excessive nitric oxide and its byproduct peroxynitrite, lipid peroxidation, apoptosis, and delayed inflammation29–32 contribute to the CO toxicity at both systemic and cellular levels.
Symptoms and Diagnosis
The symptoms predominantly manifest in organs and systems with high oxygen utilization. The severity of clinical manifestations is varied depending on the concentration of CO. For instance, the central nervous system symptoms such as headache, confusion, and collapse may occur when the blood COHb level is from 40% to 50%. Symptoms such as unconsciousness, intermittent convulsions, and respiratory failure may occur if COHb level exceeds 60%, and eventually leading to death if exposure continues. The cardiovascular manifestations result in tachycardia, increase in cardiac output, dysrhythmias, myocardial ischemia, and hypotension depending on the severity of poisoning. The correlations of clinical manifestations and severity of CO poisoning are summarized in Table 94-3.
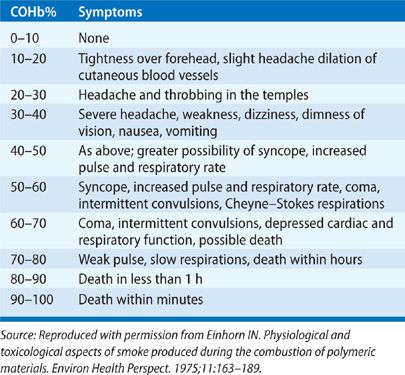
Blumenthal35 correlated the clinical signs and symptoms of CO poisoning to the concentration of the inhaled CO: headache and dizziness—up to 100 ppm, 0.01% air concentration, loss of judgment, confusion, and disorientation—up to 200 ppm, 0.02%, and nausea, vomiting, fatigue, confusion, disorientation, visual disturbance, syncope, up to the onset of coma—800 ppm, 0.08% to 12,800 ppm, 1.28%.
Diagnosis should be based on direct measurement of COHb levels in arterial or venous blood by co-oximetry. Portable breath analyzers may be used at the scene. Inability to differentiate oxyhemoglobin from COHb limits the use of pulse oximeters. Although there are some controversies on its reliability, the fingertip pulse co-oximetry, a new technology available since 2005, can be used at the scene of the fire for measurement of COHb.36 The use of blood gas analyzers that estimate SO2 based on measurement of dissolved PO2 should be avoided. The measurements of acid–base balance, plasma lactate levels, and bicarbonate are helpful in management of CO poisoning with accompanying lactic or metabolic acidosis. It is important to note that high oxygen concentrations are usually administered to the victim in transit to the hospital, and some delay from cessation of exposure to measurement of CO may limit evaluation of the true extent of exposure.37 A nomogram has been developed that can relate the COHb levels of a patient to the values that may have been present at the time of smoke inhalation; this can be used to estimate the true degree of inhalation injury.38
Treatment
The half-life of COHb is 320 minutes (adult male) in room air and ~74 minutes in a person breathing normobaric 100% oxygen at 1 atmosphere.39,40 Those values are 30% less in females.41 Therefore, all fire victims should be isolated from fire site and given 100% oxygen on route to the hospital. This allows delivery of an inspired oxygen concentration of 50% to 60%, which is usually adequate. To adequately treat a CO poisoning it is also important to establish COHb level as early as it is possible. In patients with loss of consciousness, cyanosis, or an inability to maintain the airway, 100% oxygen should be delivered via mechanical ventilation through endotracheal tube until COHb levels drop below 10% to 15%. The alternative method to rapidly decrease COHb is hyperbaric oxygen therapy (HBOT) (see Chapter 93). The HBOT allows CO to dissociate from cytochromes a and a3, and to increase PO2 despite impaired hemoglobin function.42,43 Chou et al. reported that children with CO poisoning alone who are treated with HBOT are at low risk for dying regardless of initial COHb level.44 However, there is some debate on use of HBOT especially in patients with burn injury. Because of difficulty of physiological monitoring and providing emergency procedures in small chambers, unstable hemodynamic conditions and other complications such as seizures, or aspiration of severely burned patients limit the use of HBOT.45
 HYDROGEN CYANIDE
HYDROGEN CYANIDE
Hydrogen cyanide is a colorless gas with the odor of bitter almonds. However, it is difficult to detect it at the site of the fire. Cyanide is a likely weapon for terrorists because of its notoriety, lethality, and availability. Hydrogen cyanide is produced in fires involving nitrogen-containing polymers (upholstery, furniture, nylon, wool, silk, and acrylics) and may produce rapid and lethal incapacitation of a victim at the fire source.46 Toxicity of cyanide is produced by inhibition of cellular oxygenation with resultant tissue anoxia, which is caused by reversible inhibition of cytochrome c oxidase.37 It is toxic to a number of enzyme systems. The mechanism includes combination with essential metal ions, formation of cyanohydrins with carbonyl compounds, and the sequestration of sulfur as thiocyanate. However, the main target enzyme is cytochrome c oxidase, the terminal oxidase of the respiratory chain, and involves interaction with the ferric ion of cytochrome a3.47
Symptoms and Diagnosis
Diagnosis at the fire scene may be difficult. Poisoning leads to central nervous system, respiratory, and cardiovascular dysfunction resulting from inhibition of oxidative phosphorylation. It may result in dyspnea, tachypnea, vomiting, bradycardia, hypotension, coma, and seizures. Electrocardiographic S-T segment elevation, which mimics an acute myocardial infarction, may be suggestive.48 Laboratory findings of anion gap metabolic acidosis and lacticacidemia aid in confirming the diagnosis.47 The lactic acidosis that is not rapidly responsive to oxygen therapy may be a good indicator of cyanide poisoning.38,49 Also, an elevated mixed venous saturation is suggestive of cyanide toxicity. Cyanide increases ventilation through the carotid body with peripheral chemoreceptor stimulation. Increasing ventilation may augment toxicity in the early stages. Correlations of blood cyanide concentrations with clinical symptoms are summarized in Table 94-4.
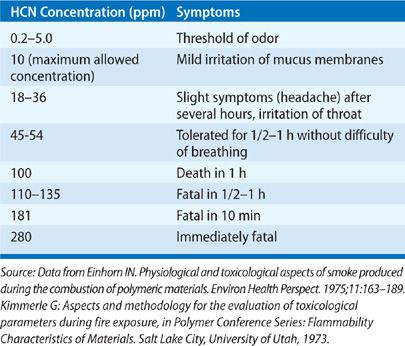
Hydrogen cyanide is found routinely in low levels in the blood of healthy individuals at levels of 0.02 μg/mL in nonsmokers and 0.04 μg/mL in smokers. Toxicity at a level of 0.1 μg/mL and at 1.0 μg/mL death is likely.51
Treatment
Fire victims suspected to have cyanide poisoning should be removed from exposure and fully decontaminated. All victims should be given pure oxygen (100%) and resuscitated properly if cardiopulmonary failure is present. Oxygen therapy appears to have strong positive effect; however, HBOT is not recommended for the reasons previously mentioned.47,51 Cyanide is metabolized by hepatic rhodanese, which catalyzes the donation of sulfur from the sulfane pool to cyanide to form nontoxic thiocyanate. The half-life time of cyanide is approximately 1 to 3 hours in humans.49,52 Although there is still controversy surrounding the treatment of cyanide poisoning, there are few antidotes available that can be used by first responders. Kelocyanor (dicobalt edetate) may be useful, but is dangerous, and requires experts to administer it.53 The following agents may be considered in an intensive care setting.
 METHEMOGLOBIN GENERATORS
METHEMOGLOBIN GENERATORS
The therapeutic goal is to convert the ferrous ion of hemoglobin to ferric ion. The resultant methemoglobin chelates cyanide to form cyanmethemoglobin (cyanide has a greater affinity for binding with methemoglobin and thus, is displaced from cytochrome oxidase). The drugs of choice in this group are sodium nitrite (intravenously) and amyl nitrite (inhaled). These drugs reduce oxygen-carrying capacity; therefore, they should be used with caution especially in patients with concomitant CO poisoning, which induces COHb that may further compromise oxygen transport. These drugs should be also used with precautions in patients with burn shock, because they are also vasodilators and can cause hypotension. In addition, there is little evidence to suggest these measures are effective, and cardiac toxicity in people with heart disease may be problematic.54 Thus, the methemoglobin generators should be used with extra precautions, carefully weighing patient conditions and comorbidities.
 SULFUR DONORS
SULFUR DONORS
The therapeutic goal is to convert cyanide to thiocyanate. The drug of choice in this group is sodium thiosulfate (intravenously). Toxicity is minimal other than an osmotic diuretic action, which may be beneficial. However, the onset of action is quite slow. Therefore, it should be used as a second-line therapy.17,55
 DIRECT BINDING AGENTS
DIRECT BINDING AGENTS
These are based on cobalt chemistry and chelate the cyanide ion directly. Hydroxocobalamin is the precursor of vitamin B12 and has very little toxicity.17,51 Minor adverse effects include transient chromaturia, alterations in renal function, reddish skin discoloration, hypertension, and rarely allergic reactions.56,57 It detoxifies cyanide by binding with it and forming cyanocobalamin, which is then excreted in the urine. The drug is currently approved for use in the United States by the Food and Drug Administration (FDA). However, there is a controversy around its efficacy.
PATHOPHYSIOLOGY
The pathophysiologic consequences of smoke inhalation on the tracheobronchial tree and lung parenchyma are discussed below.
 TRACHEOBRONCHIAL TREE
TRACHEOBRONCHIAL TREE
In normal conditions, the airway provides a protective barrier against toxic agents and bacteria, maintaining a sterile lung environment. It also facilitates airflow within the lung to ensure efficient gas exchange. Following smoke inhalation, the normal function of the airway is compromised. With rare exceptions such as inhalation of steam, injury to the airway is usually from the chemicals in smoke. The heat capacity of air is low and the bronchial circulation is very efficient in warming or cooling the airway gases so that most gases are at body temperature as they pass the glottis.58 Flames must be in almost direct contact with the airway to induce thermal injury.59 The chemicals in smoke are dependent on the materials that are being burned; however, for the most part the host response is similar. In most instances biological materials such as cotton fabric, wood, grass, or products of these such as cattle feces (commonly used as fuel in third world countries) are the fuel for the fire. These contain caustic materials such as reactive oxygen (ROS) and nitrogen species (RNS) as well as organic acids and aldehydes.60 These chemicals interact with the airway to induce an initial response to trigger an inflammatory response.
Although the degree of airway dysfunction may vary depending on the severity of inhalation injury, it can generally be characterized by few major pathologic alterations that lead to the narrowing/occlusion of the airway lumen reducing or preventing the normal alveolar gas exchange. The factors leading to the airway lumen narrowing are (1) airway hyperemia; (2) formation of an airway obstructive cast; (3) increased mucus secretion; and (4) bronchospasm (see Fig. 94-1). Airway inflammation plays a major role in the overall response to inhalation injury.
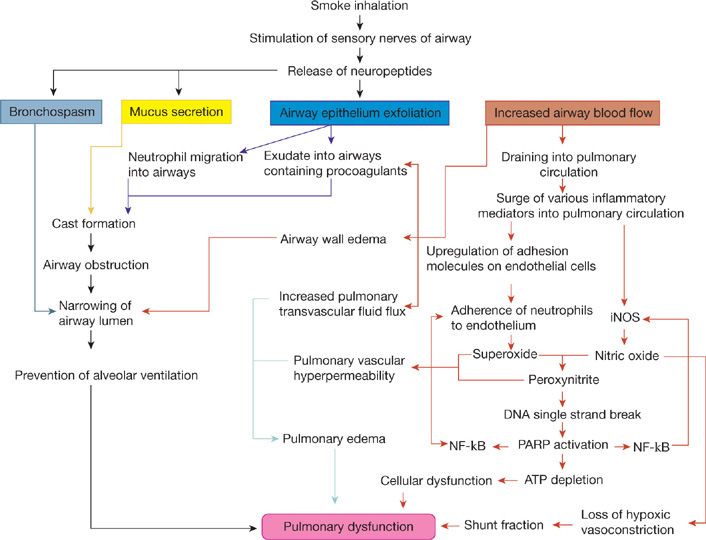
Figure 94-1 The mechanisms of lung parenchyma damage and role of airway changes following smoke inhalation injury. iNOS, inducible nitric oxide; NF-κB, nuclear factor kappa B; PARP, poly(ADP-ribose) polymerase; ATP, adenosine triphosphate.
Hyperemia of the airway is such a consistent bronchoscopic finding in smoke inhalation that it is used to diagnose the injury.61,62 Other variables that are used include injury in an enclosed space, singed nasal hair, and soot in sputum. However, these latter injuries may be present but the subject may still not develop the signs of pulmonary edema characteristic of inhalation injury.
The normal bronchial blood circulation is about 1% of cardiac output. Following the smoke inhalation, the airway blood flow rapidly increases resulting in airway wall edema and protein rich plasma leak into the airways. The data presented in Figure 94-2 confirm the dramatic (10–15 fold) increase in tracheal (A) and bronchial (B) blood flow following smoke inhalation that leads to the airway wall edema as confirmed by the macroscopic picture, which clearly illustrates the severe edema formation in the tracheal soft tissue compared to the trachea of an uninjured sheep (C). Many of the studies concerning bronchial circulation following smoke inhalation injury have been performed in sheep, because these animals have a single bronchial artery63 and a single lymphatic draining the lung that allows the measure of pulmonary transvascular fluid flux.64 Abdi et al. reported a 10-fold increase in bronchial blood flow within 20 minutes of smoke inhalation in sheep.65 These same animals demonstrate a sixfold increase in pulmonary transvascular fluid flux and a fall in PaO2/FIO2 ≤300 but these were delayed to 24 hours. Similar findings have been reported in patients with smoke inhalation alone or the combination of a large cutaneous thermal injury and smoke inhalation.66 A few investigators have also demonstrated ~10- to 20-fold increases of airway blood flow depending on the anatomical localization of the airways using the same model.67–69 These changes in blood flow are associated with increased bronchial microvascular permeability to protein and small particles70 and increased pressure.71 The ablation of the bronchial blood flow either by a ligation of the bronchial artery or the nebulization of adrenergic agonist epinephrine significantly improved the pulmonary gas exchange in the sheep smoke inhalation and burn injury model.67,69,72,73 Simultaneous with the changes in the function of the bronchial microvasculature, there is a loss or shedding of the bronchial columnar epithelium.74,75 As noted, these changes result in a perfuse transudate with a protein content similar to an ultrafiltrate of the plasma.76
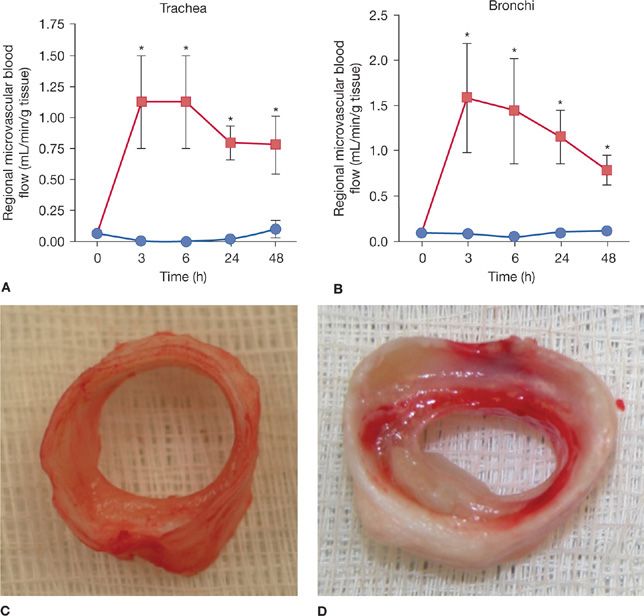
Figure 94-2 Time changes in trachea (A) and bronchial (B) blood flow in sheep following cotton smoke inhalation (48 breaths) injury. Blood flow was determined by microsphere injection technique. Data are expressed as ±SEM. *p < 0.05 versus sham. Closed circles represent sheep without injury (sham smoke), and closed squares represent sheep with smoke inhalation. C, D. Macroscopic pictures of trachea taken 48 hours after sham smoke (C) and smoke inhalation (D) in sheep.
The augmented airway circulation not only leads to the airway wall edema, but also contributes to the formation of airway obstructive casts. In conditions where the basement membrane is impaired due to smoke inhalation, the increased airway blood flow leads to the massive airway transudation. This transudate, containing procoagulants combines with copious secretions from the goblet cells and leukocytes77 to form a solid airway obstructive cast. Airway obstructive casts not only reduce/prevent normal air passage, but also become a perfect culture media for bacterial growth. In Figure 94-3, massive airway obstructive casts from human following smoke inhalation injury are shown. The airway obstructive casts, positive for submucosal gland mucus 5B (MUC5B), occupy most of the airway lumen and prevents the normal aeration of the alveoli (Fig. 94-4A and B).
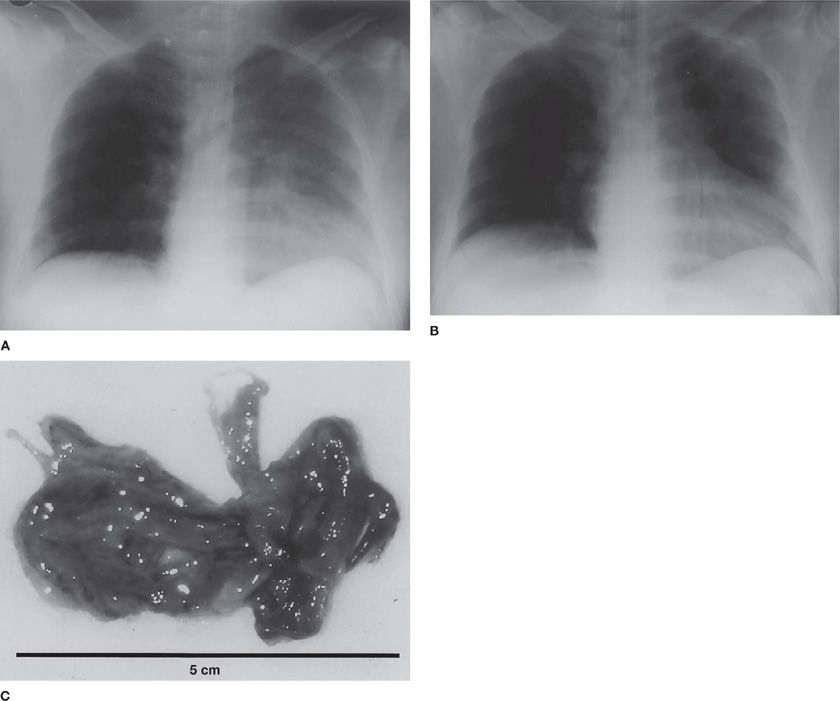
Figure 94-3 A chest X-ray of patients suffering from smoke inhalation taken at the time of admission and after removal of solid airway obstructive cast. A. On physical examination there was decreased transmission of breath sounds over the entire left chest; the chest radiograph shows obscuration of the left heart border and elevation of the left hemidiaphragm before removal of the cast. B. Radiograph taken 30 minutes after removal shows improved aeration; in addition, the left heart border is now visible. C.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


