Therapeutic Decision Making Based Upon Clinical Trials and Clinical Practice Guidelines: Introduction
Evidence-based medicine (EBM) provides a framework for informing the best possible practice. Simply, EBM aims to gather the best evidence gained from scientific investigation with the goal of optimizing medical decision making for the patients we treat. The impact of EBM is proportional to the quality and generalizability of the evidence at hand. The development of clinical practice guidelines has paralleled the growth and importance of the focus on medical quality. A discussion of the context in which practice guidelines have achieved their current prominence is followed by a presentation of their development, implementation, and maintenance. Finally, their quality and impact on medical practice are assessed.
Evidence-Based Medicine: A Building Block for Clinical Practice Guidelines
Evidence-based medicine gives the highest priority to the well-powered randomized controlled trial (RCT). The RCT is the most rigorous form of prospective scientific human experimentation to evaluate a therapeutic intervention (clinical trial) and has become the gold standard design. The RCT begins with an early stage with evaluation for safety and treating several doses (phase 1) and is followed by studies of surrogate markers of effectiveness (phase 2) and finally by definitive outcomes trials for safety and effectiveness (phase 3) (Table 13–1). It is now common to have phase 4 trials post-approval that afford the opportunity to expand the indications for a given therapy or to further explore safety concerns when events are rare but potentially fatal.2 The evidence can be best summarized by defining the patient population, the intervention, and the outcomes of interest.
| Phase | No. of Patients | Length of Phase | Goal |
|---|---|---|---|
| 1 | 20-100 | Several months | Safety, dosages, and efficacy |
| 2 | 100-500 | Several months to 2 years | Effectiveness and short-term safety |
| 3 | 500-3000 | 1-4 years | Safety and effectiveness |
| 4 | > 3000 | Ongoing | Long-term safety and rare adverse effects |
The role of bias is a critical feature of what distinguishes clinical trials from clinical practice. The most important form of bias is selection bias whereby subjects enrolled in a trial differ systematically from those individuals in the general population. In EBM, this can lead to a trade-off between internal validity (findings can be applied to the types of patients enrolled in the trial) and external validity (generalizability to a wider population). By restricting the trial cohort to particular age or medical condition criteria, trial results can be skewed against those that are underrepresented. In an analysis of 283 RCTs published in high impact general medicine journals, 72% of patients were excluded on the basis of age and 39% were excluded due to being female.3 Not surprisingly, only 47% of exclusions were judged to be strongly justified. It is common to have less than 10% of screened patients enrolled and this is due in large part to overly stringent eligibility criteria. Purists will argue that trial conclusions should be restricted to those that would have met the eligibility criteria.
Sexism has plagued clinical research from the onset. In the leading general medicine and cardiovascular journals, sex-specific results (SSRs) are consistently reported less than one-third of the time and women are consistently underrepresented in large scale RCTs (Fig. 13–1).4
Figure 13–1
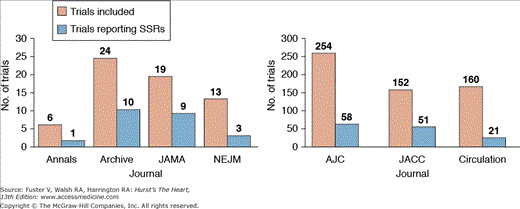
Trials reporting sex-specific results. Annals, Annals of Internal Medicine; Archive, Archives of Internal Medicine; JAMA, Journal of the American Medical Association; NEJM, The New England Journal of Medicine; AJC, American Journal of Cardiology; JACC, Journal of the American College of Cardiology. Reproduced with permission from Blauwet et al.4
Ageism is the greatest impediment to the inclusion of the elderly in clinical trials. This extends to misconceptions by both physicians and patients that older age is associated with a lower relative risk reduction for a given therapy when in fact the opposite may be true. For example, the use of thrombolytic therapy in acute ST-segment elevation MI was restricted initially to patients younger than 70 years of age. After almost 2 decades, it is now apparent that the elderly enjoy a greater net clinical benefit from this therapy when compared to younger patients. Ageism is also prohibitive since chronologic age is very different from functional age. It leads to withholding potentially efficacious therapies from deserving patients.5
Evaluating a trial population exclusively in a given race or ethnic group has also led to confusion when interpreting trial results. The LIFE trial evaluated a losartan-based to atenolol-based therapy in hypertensive patients with left ventricular hypertrophy (LVH) and found that African Americans did not respond as favorably to the losartan-based treatment as non-black patients with respect to cardiovascular outcomes (Fig. 13–2).6 In fact the hazard ratios for African Americans and non-African Americans were in opposite directions with a significant qualitative interaction (P = .016). Subsequently, the ALLHAT trial confirmed that all major antihypertensive drug classes work in all ethnic groups.7 This controversy was addressed in the A-HeFT trial which was conducted in African Americans only and showed a 43% reduction in mortality in advanced heart failure patients taking a fixed dose of isosorbide dinitrate and hydralazine versus placebo. The real question is would the trial results have been any different if whites were included?8 Secondary analyses of clinical trials should be interpreted as hypothesis generating. The effect of ethnicity, therefore, should have been prospectively tested with multiple ethnic groups involved.
Figure 13–2
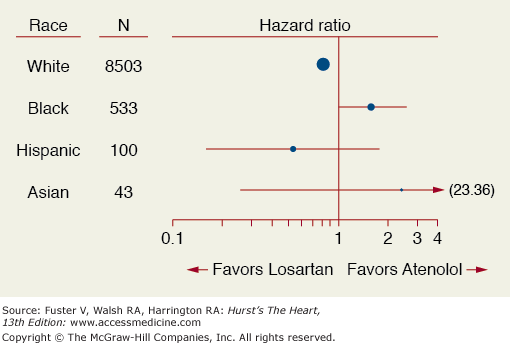
Hazard ratio for cardiovascular outcomes by race in the LIFE trial. Reproduced with permission from Julius et al.6
When considering the external validity of a trial, the first issue to address is consent bias. This is a measure of how different a study participant who has provided informed consent is from an eligible patient who has not provided informed consent. The comparison of trial with registry type patients does not address consent bias because patients in a registry have to provide only consent to be followed. Consent bias can greatly influence trial results if consenting rates are low and lead to misleading findings. Patient characteristics such as education level and ethnicity play a critical role in determining the likelihood of a patient to consent to enroll in as study. There are a number of factors that allow for the assessment of the degree of consent bias (Table 13–2).9
| Factor | Relevance | Implications |
|---|---|---|
| Does study report total number screened? | High | Essential to evaluate generalizability |
| Is the consent rate reported? | High | Response rate of 60% considered adequate |
| Is the consent method documented? | Medium | |
| Do authors address generalizability? | Medium | Acknowledges trial limitations |
| Are the baseline characteristics representative? | High | Another means of assessing generalizability |
The design of a clinical trial is an important factor in assessing the influence of bias.10 There are a number of elements that influence the robustness of the trial findings: First, is the trial double-blinded? The failure to double-blind a trial may lead to bias through the preconceived notions around the study question on the part of the trial subjects and/or trial investigators. It is estimated that failure to double-blind can exaggerate treatment effects by around 14%.11 The alternative, an open-label design, limits the conclusions that can be drawn from the trial findings. The results of the recent RE-LY trial of a novel antithrombotic agent, dabigatran compared with warfarin for the treatment of atrial fibrillation were impressive. In an open-label design, there was a 34% reduction in the risk of stroke or systemic embolism. Only future trials can confirm whether this effect size was exaggerated.12 Industry-sponsored trials are problematic if open label because of the involvement of sponsors in pre-FDA registration studies where fortunes are on the line. Investigators are often advisors and consultants to industry and have a professional stake in the trial results. The reality of conducting double-blind trials is that they are often difficult to perform and costly. On the other hand, some opponents to double-blinding argue that it creates a selection bias by restricting the trial to patients that are more educated and understand the research design implications and to investigators who might only be willing to enroll low-risk patients into such trials.
There are advances that can increase the reliability of open-label trials. The prospective, open, blinded endpoint (PROBE) design mandates an independent clinical events adjudication committee (CEC).13 The blinding of the adjudicators reduces bias and should be considered in all clinical trials. Therefore, a double-blinded design with an independent CEC provides a higher level of robustness.
Second, there has been an increasing number of trials conducted that are powered for noninferiority of newer therapies. The noninferiority comparison sets targets for the upper limit of the 95% confidence interval to fall within based upon a difference between the two arms that is predetermined. This arose from the clinical world where the question of whether an emerging therapy was no worse than the standard of care was being posed. This is particularly relevant when newer therapies may be significantly less expensive and have fewer adverse effects than the comparator. The primary limitation is that the margin of noninferiority is often ill-conceived and prone to controversy (Fig. 13–3). A recent analysis of 162 noninferiority and equivalence trials identified from MEDLINE and the Cochrane Central Registry of Controlled Trials demonstrated that only 20% provided a justification for the margins analyzed and about 21% did not specify a sample size.14 The Consolidated Standards of Reporting Trials (CONSORT) is now developing standards for non-superiority designs including non-inferiority trials to improve the quality of reporting.15
Figure 13–3
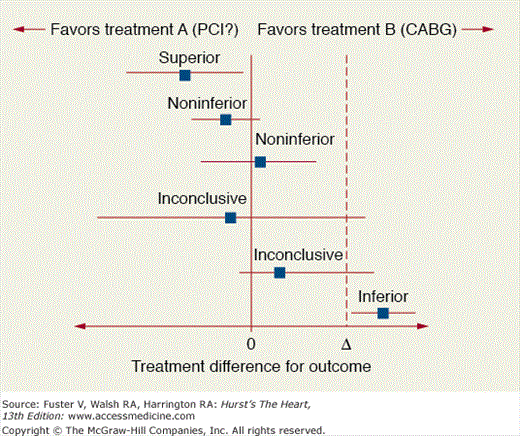
Forest plots comparisons of outcomes in noninferiority trials. Reproduced with permission from Piaggio et al.15
Third, the early stopping of trials is controversial particularly when event rates are low. Trials with small numbers of events and very large treatment effects should be interpreted with skepticism since there is a high likelihood that this effect is not true. The most troubling aspect of such trials that are stopped early is that often there is no accounting for how the decision to stop was undertaken.16,17
There is a large body of literature evaluating the relative virtues of observational studies compared to RCTs. The argument for the validity of well-conducted observational studies is strong since they can include a wider range of patients and endpoints and are, therefore, more reflective of the “real world.“
One of the most explored areas for comparing observational studies with RCTs comes from the evaluation of percutaneous coronary intervention (PCI) and coronary bypass surgery (CABG). The BARI trial demonstrated a clinical benefit of CABG over PCI in diabetic patients with multivessel disease. This led to an NHLBI alert recommending that diabetic patients meeting the BARI eligibility criteria be considered first for surgery. Multiple registries including one from the BARI investigators showed that patients subjected to a revascularization strategy free of randomization experienced similar mortality rates regardless of whether PCI or CABG was undertaken. A recent meta-analysis of long-term follow-up of the earlier RCTs also demonstrated no survival advantage for CABG. This brings into question whether the results from single well-conducted trials alone can provide definitive evidence that may be generalized to the overall population.18 The greatest challenge for observational studies is the validity of risk adjustment since, as in the example above, surgical patients are often sicker with more extensive coronary heart disease.
RCTs provide the highest level of evidence and should remain the gold-standard for evaluating therapies. The real concern with the field is that with increasingly complex trial designs, active comparator trials, and the tendency to limit follow-up of patients to shorter durations, the gap between the robustness of RCTs and observational studies may be closing. The only solution is to make it easier to perform RCTs and include a wider range of patients.
The importance of clinical judgment, the process by which clinicians make decisions about the best therapy for their patients by taking more than the evidence only into account, is minimized in clinical trials. Many interventional cardiologists believe that this explains the BARI registry results. Instead of a “winner takes all“ philosophy that surrounds superiority randomized trials, the interventionalists are willing to accept that higher risk patients may indeed fare better with bypass surgery.
The importance of co-intervention cannot be exaggerated. The background therapy administered to all arms of a randomized trial directly impacts the event rate and therefore the feasibility of the study. In clinical practice, patients are often receiving different background therapy than in a trial due to less adherence to guidelines and other factors including cost of these therapies.
In the last decade, clinical trials have begun recruiting in developing countries such as India and countries with less advanced health care systems such as the Ukraine and China. This has resulted in faster recruitment at a lower cost since recruitment rates are higher and subsequently trial duration is reduced considerably. Physician-investigators can earn a considerable proportion of their annual income by participating in lucrative industry-sponsored trials.19
The downside of these global trials is that co-interventions such as background medical therapy or interventions with devices are less prevalent because of inaccessibility and economics. This also often involves less access to proven therapies that reduce the baseline risk profile. Many patients may be treatment naïve, which may affect the performance of a specific compound.
One of the most important aspects of clinical trials is selection of the primary outcome measures. This often shapes how influential the trial is in changing practice down the road. Recently, the PCI versus CABG question for treatment of coronary disease was addressed by a large RCT, SYNTAX. The trial investigators chose a 1-year end point as opposed to a longer one such as the 5-year end point in BARI, and found that PCI with a drug-eluting stent strategy was not non-inferior to CABG. The major controversy is that in order to be powered for 1-year outcomes, repeat revascularization was included along with hard cardiovascular endpoints in the primary outcome cluster. Each individual event-type was weighed equally. The difference between the two arms was totally explained by the difference in the rate of repeat revascularization.
One of the most misinterpreted parts of clinical trials is the measure of clinical benefit. Commonly, the findings of RCTs are expressed as relative risk or relative risk reduction. Relative risk reduction (RRR) usually remains durable across the spectrum of baseline risk but is misleading when event rates are low. The number needed to treat (NNT), which is clinically more useful, is the inverse of the absolute risk reduction times 100 and has been found to influence clinical decision making to a greater extent than RRR. Number needed to treat is rarely used as the primary measure of benefit in clinical trials despite affording the best opportunity to weigh risks and benefits. Therefore, the result of clinical trials in populations of patients needs to be expressed in a way that can be useful to clinicians. The confusion that results can lead to errors in clinical decision making.
All too often, trials may be underpowered to detect small yet clinically important differences. The dilemma of the negative trial leads to the false conclusion of no difference between therapies when in fact one exists.
Clinical trials often fall short of adequately estimating safety concerns that are rare, since they are often significantly underpowered to detect such risks. The Cochrane Collaboration has shown that, of the 38 systematic reviews with more than 4000 patients randomized, greater than 50% had no data regarding harm of the intervention under evaluation. A good example of the challenges of assessing safety when events are relatively rare comes from the COX-2 inhibitor field, where there was a signal of increased cardiovascular risk with rofecoxib. Because the large-scale outcomes trial was conducted using the higher 50-mg dose, it was not until the large database analyses that the dose-response toxicity was apparent. Perhaps rofecoxib would still be on the market if the 25-mg dose had been studied. An extension of the CONSORT statement on harms recommends improvements on reporting.20 A means of linking phase 3 clinical trials with phase 4 postmarketing surveillance would be a step in the right direction. The FDA has made great strides in identifying the challenges of late pharmacovigilance. The FDA Amendments Act provides a framework to create such surveillance. The recent controversy of the safety of antidiabetic medications will likely be addressed through this mechanism.21
In the past, there has been great concern that only a fraction of trials that are completed actually are eventually published. This has led to a serious form of bias termed publication bias.
The selective publication of antidepressant trials has been evaluated.22 Using FDA data on 12 antidepressant drugs, Turner and colleagues discovered that 31% of trials were not published. This had a marked effect on how the trials were interpreted. Using searches of published trials, 94% were considered to have a positive result, whereas, the FDA data shows that only 51% were positive.
Recently, the ICMJE has instituted a strategy that only trials that have been preregistered will be published in their member journals.23,24 This was a direct response to the observation that if researchers did not register their trial there would be no obligation to report their findings, especially if the results were negative or unfavorable for a newer therapy.25 As well, it raised the standard for having to follow a prescribed protocol that was declared a priori to the final results. Registration on ClinicalTrials.gov has been widely accepted as the gold standard and is now a requirement for any serious investigation. These repositories are now including a wide spectrum of clinical trials including phase 1 trials.26
The reporting of trial results has often been of poor quality and prone to misinterpretation. In response to these observations, the CONSORT group established a 22-item checklist that standardizes trial reporting.
The importance of generalizing findings from clinical trials is realized one patient at a time. For clinicians doing their best to practice evidence-based medicine the real question is does this finding apply to the individual patient in front of me?27,28 Because of the emerging evidence that shows wide individual variations in the response to given therapies, conventional sub group analyses of RCTs are often inadequate to detect these variations. A good example of variations in response to therapies comes from the antiplatelet world. In the Optimizing Antiplatelet Therapy in Diabetes Mellitus (OPTIMUS) trial, about two-thirds of the diabetic patients with established coronary heart disease and on a stable dose of aspirin and clopidogrel 75 mg daily were found to be hyporesponders to clopidogrel.29
There has been a trend toward pooling of data at either published level or patient level to summarize treatment effects in cardiovascular medicine. The advantages to meta-analysis is that it provides robust information on the direction of effect and helps to understand these effects in important sub groups such as diabetic elderly patients. There, however, have been many instances where the quality of meta-analysis is poor and unlikely to provide any important contribution.30,31
Clinical trials by their very nature do not capture the patient’s role in decision making and, therefore, will always be limited in the capacity to change actual clinical practice.32 This is where clinical judgment, by encompassing clinician experience, will always involve more than the evidence itself.33
The application of findings from clinical trials to our daily practice is challenging and must always be put into the context of the uniqueness of each patient. The more inclusive the eligibility criteria and the more rigorous the trial design, the more likely that these findings are valid and generalizable to real-world patients.
Clinical Practice Guidelines
In 1989, the Agency for Health Care Policy and Research (AHCPR, since renamed the Agency for Healthcare Research and Quality [AHRQ]) was created with the charge to “enhance the quality, appropriateness, and effectiveness of health care services, through the establishment of a broad base of scientific research and through the promotion of improvements in clinical practice and in the organization, financing and delivery of health care services.“ Specifically included in the legislation was the charge that the agency put forth “clinically relevant“ practice guidelines. The Institute of Medicine convened an advisory committee at the time to assist the newly formed agency in fulfilling its mandate. The committee’s report defined practice guidelines as “systematically developed statements to assist practitioner and patient decisions about appropriate health care for specific clinical circumstances.“ 34 The intended utility of practice guidelines was expressed in a follow-up report by the Institute of Medicine in 1992: “Scientific evidence and clinical judgment can be systematically combined to produce clinically valid, operational recommendations for appropriate care that can and will be used to persuade clinicians, patients, and others to change their practices in ways that lead to better health outcomes and lower health care costs.“ 35 Although the report acknowledged the existence of substantial barriers to the realization of this ideal, it remains a concise statement of the definition and promise of practice guidelines.



