Chapter 1
The Vascular Endothelium in Diabetes
Introduction
The vascular endothelium, the monolayer of thin cells lining the arteries and veins, serves as the key regulator of arterial homeostasis. It plays a vital role in regulating vascular tone, cellular adhesion, platelet activity, vessel wall inflammation, angiogenesis, and vascular smooth muscle cell proliferation. In order to regulate these functions, a number of important vasoactive molecules, including nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), prostacyclin (PGI2), and endothelin (ET-1), are produced and released by the endothelial cells [1, 2].
Normal Endothelial Cell Function
The arterial endothelium is composed of a layer of spindle-shaped endothelial cells that are bound together by tight junctions and communicate directly with each other and the underlying smooth muscle cells via gap junctions. This forms a protective barrier between the blood and the rest of the vessel wall that is relatively impermeable to low-density lipoprotein (the core component of atherosclerotic lesions), able to sense molecular cues and interact with cellular components of the circulating blood.
Furchgott and Zawadzki first demonstrated in 1980 that endothelial cells are essential in order for underlying smooth muscle relaxation to occur in response to acetylcholine administration in the rabbit aorta [3] and NO was subsequently identified as this endothelium-derived relaxing factor [4]. A healthy endothelium is able to secrete NO, a diatomic molecule generated from L-arginine, by the action of the enzyme endothelial NO synthase (eNOS) in the presence of cofactors such as tetrahydrobiopterin [5]. NO exerts its action by diffusing into vascular smooth muscle cells where it activates G-protein-bound guanylate cyclase, resulting in c-GMP generation, smooth muscle relaxation, and vasodilatation [1] (Figure 1.1). eNOS, in normal physiology, is activated by shear stress from blood flow through the vessels and also by molecules such as adenosine, bradykinin, serotonin (in response to platelet aggregation), and vascular endothelial growth factor (induced by hypoxia; Figure 1.1) [6–8].
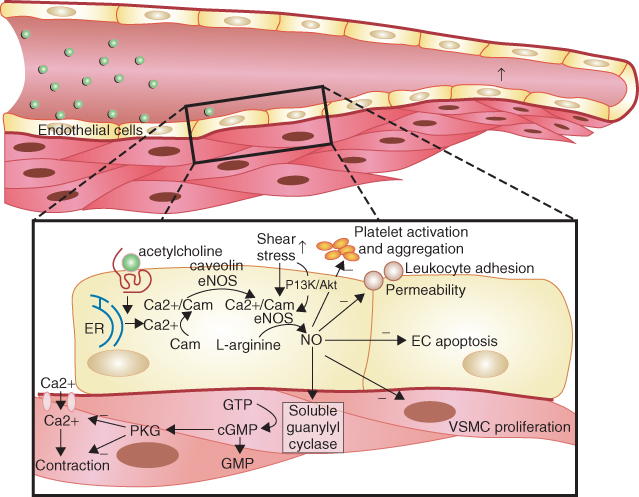
Figure 1.1 Illustration of the stimulation of endothelial NO synthase by acetylcholine and shear stress leading to increased nitric oxide (NO) production in endothelial cells by receptor and nonreceptor and calcium-dependent and noncalcium-dependent pathways. (Source: Herrmann J et al. 2010 [8]. Reproduced with permission of Oxford University Press.)
In addition, NO has antiplatelet effects and can down-regulate inflammatory pathways and also decrease the generation of ET-1, a potent vasoconstrictor polypeptide, which also possesses pro-inflammatory, pro-oxidant, and pro-proliferative activity [9].
Other endothelial-derived vasodilators exist and act independently of NO to maintain vasodilator tone. PGI2, produced from the cyclooxygenase system, and EDHF are such molecules, with the latter able to compensate for the loss of NO-mediated vasodilator tone when NO bioavailability is reduced [10, 11]. Normal health and physiological functioning of the vascular endothelium are maintained by a balanced release of endothelial-derived relaxing factors, such as NO and prostacyclin (PGI2), and vasoconstricting factors like ET-1 and angiotensin II. The dysequilibrium of their production, release, and action is the chief characteristic of endothelial dysfunction [12].
Beyond its function in regulating vessel tone, the vascular endothelium also serves to play an important role in both mediating and responding to inflammatory pathways. In addition to its constrictor effects, angiotensin II generated by the endothelium has effects on vascular smooth muscle cell contraction, growth, proliferation, and differentiation. A range of selectins and adhesion molecules are produced, resulting in the binding and transendothelial migration of inflammatory cells [13, 14]. Furthermore, the endothelium is directly involved in the balance between coagulation and fibrinolysis, which is mediated by its synthesis of both tissue-type plasminogen activator (t-PA) and its inhibitor, plasminogen activator inhibitor-1 (PAI-1) [12, 15].
Measuring Endothelial Function
Following the in vitro work of Furchgott and Zawadzki, Ludmer et al. demonstrated for the first time in humans that locally administered acetylcholine caused vasoconstriction of atherosclerotic coronary arteries and vasodilatation in normal coronary vessels in subjects undergoing cardiac catheterization [16]. Subsequently, a noninvasive method was developed for assessing endothelial function in the conduit arteries of the peripheral circulation. This method used a period of forearm ischemia followed by reactive hyperemia to increase blood flow through the brachial artery, increasing local shear stress, mediating NO release and brachial artery dilatation [17]. Peripheral endothelial vasodilator function correlates with coronary endothelial function and cardiovascular risk factors, including smoking, dyslipidemia, and diabetes, and can predict incident cardiovascular events in older adults [18–21].
Various techniques have been developed that use pharmacologic agents to act on the endothelium or that measure the vasodilator response to increased shear stress. No one test has been shown to be ideal and indeed a combination may be required to evaluate fully the various aspects of vascular endothelial biology (Figure 1.2).
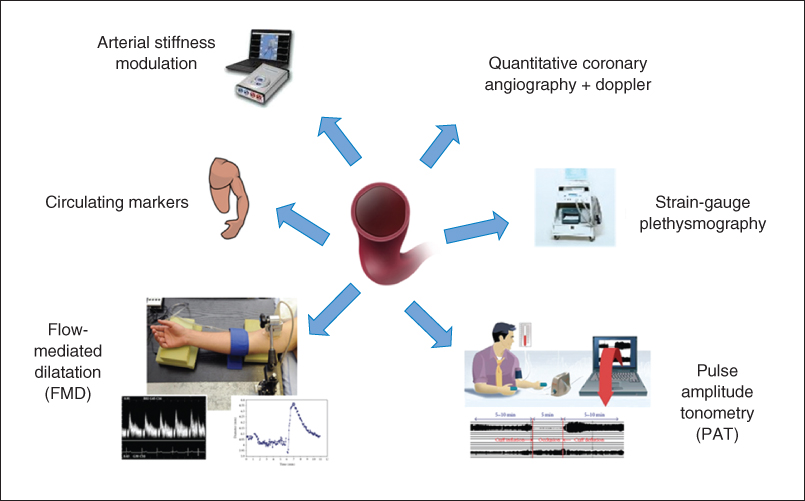
Figure 1.2 Methods for assessing human endothelial function.
Invasive methods for assessing endothelial function include venous occlusion plethysmography and quantitative coronary angiography with Doppler flow wire to assess coronary diameter and blood flow.
The original tests of endothelial function used the latter techniques to assess coronary circulatory physiology. Pharmacologic agents, such as acetylcholine, are used to induce an endothelium-dependent vasomotor response, measuring changes in the epicardial and microvascular circulation. At the doses traditionally used, a vasodilator response is usually observed in normal coronary vessels, but in the presence of endothelial dysfunction, where NO bioavailability is reduced, the action of acetylcholine on smooth muscle muscarinic receptors predominates, resulting in vasoconstriction [22]. This method of measuring endothelial function is limited to patients with more advanced and established arterial disease who warrant cardiac catheterization, but is helpful in quantifying the response to potential beneficial therapeutic agents, such as statins, on endothelial function [23].
A further invasive technique for evaluation of forearm microcirculation and resistance is by measuring changes in forearm blood flow (FBF) using venous occlusion strain-gauge plethysmography [24]. The method uses the contralateral arm as its control, with most studies assessing percentage differences in FBF and vascular resistance between experimental and control arm after the administration of endothelium-dependent and endothelium-independent agonists. By using eNOS antagonists, such as L-NMMA, the contribution of NO to vasomotor regulation can be inferred; the technique can also be used in healthy controls and allows other vasomotor pathways to be studied in detail. Its invasive nature, however, thus limits its use to smaller studies and its clinical relevance to conduit vessel atherosclerosis is also questioned.
The noninvasive methods of measuring endothelial function are inherently more practical in that they can be more readily used in large patient groups. Flow-mediated dilatation (FMD) using ultrasound stands as the current gold-standard technique for noninvasive assessment of endothelial function. The rationale is based on the reactive blood flow in the brachial artery following a five-minute period of forearm ischemia caused by suprasystolic inflation of a blood-pressure cuff. The increased shear stress during the resulting hyperemia stimulates NO release from the endothelium, causing smooth muscle relaxation and dilatation of the artery. By imaging the brachial artery with high-resolution 2D ultrasound and using pulsed-wave Doppler interrogation, changes in arterial diameter and blood flow can be assessed [17]. When care is paid to methodology, FMD has been demonstrated to have good reproducibility [25]. Some differences in techniques, including cuff position and duration of cuff occlusion, remain areas of controversy in using this method [26–28], although guidelines have been produced in an attempt to reduce the variability of the methodology in research [29, 30]. Despite variations in methodology, FMD stands as a reliable method of measuring endothelial function and is associated with coronary endothelial vasodilator function and circulating markers of endothelial activation, as well as being a predictor of long-term cardiovascular outcomes [21, 31].
Another useful noninvasive technique that is emerging for measuring endothelial function is pulse amplitude tonometry (PAT). The same stimulus as FMD is used and the EndoPAT system employs a probe placed on the fingertip to record changes in arterial pulsatile volume. Both fingertips are used for recordings in order to have an internal control. Measurements are made at baseline and following reactive hyperemia (RH) so as to allow an RH-PAT index (ratio) to be calculated. The RH-PAT signal is decreased with risk factor expression, has been shown to correlate well with risk factor burden, and can help to identify coronary vascular dysfunction [32, 33]. Reproducibility has been shown to be similar to that of FMD. Although the mechanism of vasodilatation is not entirely NO dependent and the autonomic nervous system may also have an influence on the fingertip pulse waveform [34], RH-PAT is widely considered to be a useful and practical tool for assessing endothelial dysfunction.
Endothelial function can also be assessed using pulse wave velocity (PWV) measurement. This method measures the speed of transit of the arterial pulse-pressure waveform through an artery, thus providing information on arterial stiffness and endothelial function. A similar protocol to that of FMD, with RH stimulus, has been devised by Naka et al. involving placing one cuff at the wrist and one on the upper arm, with RH induced following the occlusion of the wrist cuff. The subsequent NO release and reduction in arterial tone cause a slowing in PWV, reflecting the magnitude of endothelial NO release [35].
Although these newer methods, particularly RH-PAT, appear promising in their use for assessing endothelial function, FMD currently remains the technique of choice and has become widely used in clinical studies.
Circulating Markers of Endothelial Dysfunction
In addition to invasive and noninvasive methods of assessing endothelial function, such as coronary angiography or FMD, there are a number of circulating biomarkers that reflect the degree of endothelial activation and dysfunction (Table 1.1).
Table 1.1 Circulating biomarkers of endothelial function.
| Biomarkers |
| Nitric oxide Nitrite ion Asymmetric dimethyl arginine Endothelin-1 Interleukins Chemokines Adhesion molecules (VCAM-1, ICAM-1) Selectins (E-selectin, P-selectin) Plasminogen activator inhibitor- 1 Tissue plasminogen activator Von Willebrand factor Endothelial microparticles microRNAs Circulating endothelial cells Endothelial progenitor cells Endothelial microparticles |
Given that endothelial activation and dysfunction are characterized by the change in the balance of vasomotor factors released by the endothelium, measuring circulating markers and mediators of this dysfunction have been shown to provide important pathological insights into the influence of the endothelium on atherosclerotic disease, although the systemic levels of these markers may not necessarily represent their true local effects on the vascular wall.
Endothelial activation results in vascular inflammation. Thus, an array of inflammatory cytokines, adhesion molecules, regulators of thrombosis, measures of NO biology, as well as markers of endothelial damage and repair can be evaluated to inform on these processes. These measures can be helpful markers of the severity of endothelial activation and dysfunction in a population and can complement other physiological tests of measuring endothelial function [36].
No precise circulating marker reflecting local and systemic generation of NO is available, although levels of nitrite and nitrate have been suggested as indirect measures. Asymmetric dimethylarginine (ADMA), an endogenously derived competitive antagonist of eNOS, is quantifiable; higher levels are typically present in those patients with cardiovascular risk factors, such as dyslipidemia and diabetes, and may contribute to the endothelial dysfunction. Higher levels of ADMA have been associated with reduced NO bioavailability in animal and clinical studies [37, 38]. Logistical and financial barriers currently preclude its use in routine clinical practice.
The inflammatory cytokines and adhesion molecules generated by endothelial activation, reflecting the stimuli to leucocyte migration into the subendothelium, can also be measured. Vascular cell adhesion molecule 1, intracellular adhesion molecule 1, and E- and P-selectins are examples, with E-selectin most specific for vascular endothelial activation. Circulating levels of such molecules are typically associated with adverse cardiovascular outcomes [39, 40].
In addition, MicroRNAs (miRNAs), a group of noncoding small RNAs, are emerging as important molecules in endothelial dysfunction in diabetes and may indeed shed light on these underlying disease processes. In the hyperglycemic environment, for example, miRNAs decrease endothelial cell proliferation and migration, as well as causing cell cycle inhibition, resulting in vascular endothelial dysfunction [41]. As levels of miRNA in the serum of humans have been shown to be stable, reproducible, and consistent among healthy individuals, it is thought they may become clinically useful biomarkers of vascular status in patients with diabetes [42, 43].
Similarly, markers of a prothrombotic state can be measured, which may reflect endothelial damage and activation; for example, the change in the balance of tissue plasminogen activator and its endogenous inhibitor, plasminogen activation inhibitor-1 [44].
As measures of endothelial cell injury and repair are a reflection of endothelial activation and dysfunction in the disease process, assays have been developed to examine the detachment of mature endothelial cells and microparticles derived from activated endothelial cells, reflecting damage, and the number and characteristics of circulating endothelial progenitor cells (EPC), reflecting repair. Assessment of the relationships between these populations can shed light on the balance between injury and repair (in diabetes) that may have a future role in clinical practice and in risk assessment of high-risk patients [45]. Endothelial microparticles (EMP) result from endothelial plasma membrane blebbing and carry endothelial proteins such as vascular endothelial cadherin, intercellular cell adhesion molecule (ICAM)-1, E-selectin, and eNOS [46–48]. Their shedding from activated or apoptotic endothelial cells reflects their role in coagulation, inflammation, endothelial function, and vascular homeostasis. The exact role of EMP in vascular homeostasis remains unclear. There is evidence that they can actually promote cell survival and induce endothelial regeneration [49] and, although promotion of angiogenic processes by EMP may have beneficial effects in ischemia, this could be detrimental for plaque stability and in proliferative diabetic retinopathy [50]. In diabetes it has been shown that higher levels of EMP are associated with endothelial activation and apoptosis [1, 51]. Furthermore, interventions to treat patients with type 2 diabetes with calcium channel blockers have shown decreases in EMP, suggesting the latter’s potential use as biomarkers of vascular endothelial dysfunction in diabetes, although their specific clinical utility remains to be defined [52, 53].
Endothelial Cell Dysfunction
Endothelial dysfunction results from a loss of the homeostatic balance between endothelial-derived relaxing factors, such as NO, and contracting factors, such as ET-1. A number of cardiovascular risk factors have been implicated including dyslipidemia, diabetes mellitus, hypertension, and smoking. In these circumstances, the endothelium is activated, with an increased expression of leucocyte adhesion molecules, release of cytokines, and inflammatory molecules. The resulting inflammation and arterial damage continue in a self-promoting fashion, contributing to the initiation and development of atherosclerotic plaque formation and its clinical consequences such as myocardial ischemia or infarction [54, 55].
One of the defining characteristics of endothelial activation is reduced NO bioavailability. This largely occurs in the context of increased oxidative stress, when the enzyme, eNOS, may switch to generate superoxide (reactive oxygen species or ROS), a process known as “eNOS uncoupling.” This is thought to occur when the key cofactor tetrahydrobiopterin is not present or when the substrate, L-arginine, is deficient [56]. In addition, ROS, in the presence of superoxide dismutase, leads to the production of hydrogen peroxide. These molecules can target cellular regulatory proteins, such as NFκB and phosphatases, promoting inflammatory gene transcription [1, 57]. The mitochondrion is thought to be an important source of ROS in which the production of free radicals and mitochondrial superoxide dismutase capacity is carefully regulated during physiological cellular homeostasis. During hypoxia, or in disease processes with increased substrate, such as obesity and type 2 diabetes with hyperglycemia and increased free fatty acids, this fine balance can be disturbed, resulting in increased free radical generation. Xanthine oxidase and NADPH oxidase are other important sources of oxidative stress in the endothelium, with xanthine oxidase activity having been shown to be increased by over 200% in patients with coronary artery disease compared with controls [58].
A further effect of prolonged exposure to cardiovascular risk factors is the effect on endothelial damage and repair. Normal endothelial integrity depends on its ability to repair and on any degree of localized injury. Endothelial cells are able to replicate locally to replace injured and lost cells, but also EPC recruited from the bone marrow circulate and are able to home to areas of injury and promote local repair processes in the endothelium [59–61]. It is known that eNOS is important in the regulation and function of EPC [62], that decreased levels of EPC are correlated with increased risk of coronary artery disease [63, 64], and that interventions, such as statin therapy, increase EPC in high-risk patients, including those with coronary artery disease [65]. In diabetes it has been shown that levels of EPC and circulating angiogenic cells (CAC) are reduced in relation to smooth muscle progenitor cells (SMPC), reflecting damage; this may therefore translate into reduced vascular repair capacity and promote macrovascular disease in type 2 diabetes [66]. The reduction in EPC in diabetes may also explain the pathogenesis of microangiopathy, as clinically significant correlations have been found in nephropathy and retinopathy [67, 68]. Furthermore, in diabetes EPC have functional defects such as impaired proliferation and adhesion, which are also likely to be of importance [69, 70]. Thus EPC are thought to play an important role in maintaining normal vascular endothelial function in diabetes.
Endothelial Cell Dysfunction in Diabetes
Both micro- and macrovascular complications are the major causes of morbidity and mortality in patients with diabetes, and endothelial cell dysfunction is believed to be pivotal in the development of associated vascular injury. There are a number of factors specific to diabetes that contribute to endothelial dysfunction (Figure 1.3).
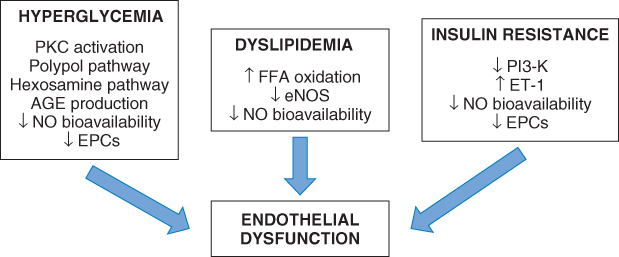
Figure 1.3 Mechanisms increasing oxidative stress and resulting in endothelial dysfunction in diabetes.
Hyperglycemia
Hyperglycemia in both type I and type II diabetes has been implicated in the pathogenesis of microvascular complications in large clinical trials [71–73].
Oxidative stress in endothelial dysfunction in diabetes is chiefly driven by hyperglycemia. The high glucose levels up-regulate the polypol pathway, which usually converts excess intracellular glucose into sugar alcohols by the enzyme aldose reductase. Normally, very little glucose is utilized by this pathway. In diabetes, an overproduction of ROS by the mitochondrion leads to increased aldose reductase activation, with conversion of glucose to sorbitol and then oxidation to fructose. This results in increased ROS production, subsequent inactivation of NO, and inhibition of endothelium-dependent dilatation [74–76]. Intracellular hyperglycemia also activates the hexosamine pathway, resulting in increased expression of PAI-1 [77].
Furthermore, high levels of intracellular glucose activate the enzyme protein kinase C (PKC), which can result in overexpression of the fibrinolytic inhibitor PAI-1 and activation of NFκB in endothelial cells, resulting in an increased propensity to thrombotic and atherogenic occlusion and further inflammation [78, 79]. In addition, PKC activation by hyperglycemia can cause increased vascular permeability and angiogenesis via increased expression of vascular endothelial growth factor (VEGF) in endothelial and smooth muscle cells [80].
Hyperglycemia is also causally implicated in the production of AGE, the circulating and intracellular proteins that have undergone nonenzymatic glycation. AGE have been linked to vascular inflammation, dysfunction, and injury through various mechanisms, including overproduction of ROS [81]. The main mechanism is through the binding of AGE to their receptors (RAGE), resulting in activation of NFκB and generation of ROS [76, 82, 83]. Raised plasma levels of endogenous RAGE have been noted in patients with type 2 diabetes and nephropathy [84].
It is through increased oxidative stress, as well as increased intracellular calcium, mitochondrial dysfunction, and changes in intracellular fatty acid metabolism, that hyperglycemia is thought to result in endothelial cell apoptosis [12].
In addition to its influence on oxidative stress and AGE production, hyperglycemia has also been associated with decreased NO bioavailabilty. Kawano et al. showed that hyperglycemia rapidly suppresses flow-mediated endothelium-dependent vasodilatation of the brachial artery [85]. Furthermore, in studies of human umbilical vein endothelial cells, it has been shown that elevated glucose inhibits NO production [86]. In contrast, some studies have demonstrated that NO release is increased in hyperglycemic conditions, with eNOS activity increased in the cardiac endothelium of rats with diabetes [87], leading to the suggestion that eNOS uncoupling may actually occur secondary to hyperglycemia of diabetes and explain endothelial dysfunction [88, 89]. Furthermore, endothelial dysfunction in diabetes is also related to an increase of endothelial-derived constricting factors (EDCFs), likely secondary to exposure of the endothelial cells to high glucose, causing oxidative stress and overexpression of COX-1 and COX-2, and thus involvement of COX-derived prostanoids [90]. ET-1 is also known to be present in higher levels in patients with type 2 diabetes compared with healthy subjects, and this is accompanied by increased oxidative stress and proinflammatory markers [91].
Finally, it is worth noting that the severity of hyperglycemia, as measured by HbA1c, in both type 1 and type 2 diabetes, correlates with lower levels of circulating EPC, resulting from either impaired proliferation, reduced mobilization from the bone marrow, or shorter circulating time [92, 93]. This has the potential consequence of reducing the vascular repair capacity in diabetes.
It should be noted, however, that despite evidence for hyperglycemia being responsible for these mechanisms leading to endothelial cell dysfunction, some evidence points toward endothelial dysfunction preceding marked hyperglycemia in diabetes. For example, the nonobese diabetic (NOD) mouse model for type 1 diabetes has shown that endothelial dysfunction is present, with evidence of vasoconstriction, prior to development of hyperglycemia [88]. Therefore, the cause and consequence of hyperglycemia and endothelial dysfunction may not be so obvious.
Insulin Resistance
Although hyperglycemia is common to all types of diabetes, insulin resistance is more a characteristic of type 2 diabetes and its role in endothelial cell dysfunction is important.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


