CHAPTER 45 The Use of Genetic Science in Thoracic Disease
MECHANISMS OF CARCINOGENESIS AND METASTASIS
Progression of normal bronchial or esophageal epithelium to overt carcinoma is the result of a series of acquired genetic alterations that are under the influence of multiple environmental, biologic, and molecular processes. Non–small cell lung cancer (NSCLC) illustrates many features of this complex relationship. NSCLC pathogenesis is clearly influenced by carcinogens such as those in tobacco smoke. The critical control points in this sequence of malignant degeneration include various proto-oncogenes and tumor suppressor genes. Several studies have demonstrated that genetic abnormalities occur early in the process and remain persistent even after cessation of smoking or even with more limited exposure to carcinogen.1,2 In fact, the development of tumors may occur with either the activation or the deletion of those critical regulatory genes. A number of genetic abnormalities have been identified frequently in early-stage lung cancers, such as mutations in the p53 tumor suppressor gene, mutations in the K-ras proto-oncogene, hypermethylation of the p16 tumor suppressor gene promoter, and loss of heterozygosity in chromosomal regions of importance.1 Those genetic events set the stage for the development of metastatic deposits of tumor.
Tumor metastasis is an equally complex series of molecular events. The fundamental steps required for metastasis include ongoing local tumor proliferation, angiogenesis,3,4 invasion, dissemination, and implantation.5,6 According to the prevailing model of tumor dissemination, this capability is acquired relatively late in a stepwise process of tumor progression. Initially, certain cells of the nascent tumor with advantageous growth characteristics are further selected to become the progenitors of successor cells that may predominate the tumor mass. These advantageous phenotypes must include self-sufficiency in growth signals, resistance to antigrowth signals, limitless replication capability, sustained angiogenesis, and evasion of apoptosis.7 Subsequently, individual cells in these large populations acquire additional unique mutations conferring the rare capability to undergo the metastatic cascade. Only a subset of cells in the primary tumor will attain this phenotype after successive genetic aberrations.8,9 Thus, any primary tumor mass comprises heterogeneous cell populations diverse in genotype and biologic behavior. At the molecular level, a number of genes have been implicated in each of the distinct steps of metastasis, but the precise mechanisms remain incompletely understood. An increasing body of evidence supports the idea that a tumor has the capacity to grow from a small number of cells that have the ability to self-renew and differentiate. These cells are termed cancer stem cells. Even this general understanding of carcinogenesis and metastasis can lead to rational approaches that help us develop and interpret the genetic analysis of thoracic malignancies.
MOLECULAR MARKERS: CURRENT CONCEPTS
DNA-based markers include single nucleotide polymorphisms (SNPs), chromosomal translocations, changes in DNA copy number, DNA microsatellite instability, and differential promoter methylation. In addition to these DNA aberrations, mutations in oncogenes, tumor suppressor genes, and mismatch repair genes can occur. Identification of these altered genes may yield a molecular marker that can be investigated and potentially used clinically. For example, several oncogenes with mutations are known to be predictive of prognosis in patients with NSCLC (K-ras, p53, cyclin-dependent kinase inhibitor A, retinoblastoma gene).10–16 MYC (2.5-10%), cyclin D1 (5%), and epidermal growth factor receptor (EGFR) (6%) are amplified and overexpressed in patients with NSCLC. C-erbB2 or BCL2 overexpression is involved in 25% of cases.17 Other important considerations at the DNA level include the epigenetic regulation of transcription and translation, which may lead to carcinogenesis. Recently, gene silencing by CpG methylation has received a fair amount of attention: identifying hypermethylated DNA may help characterize malignancy in the sputum or in bronchoalveolar lavage fluid. SNPs are another area of intense investigation for human lung cancer. A recent study of the 250K SNP array from Affymetrix (Santa Clara, CA) focused on evaluating copy number changes in 371 adenocarcinomas of the lung. Of the 57 recurrent alterations found, many had not been previously discovered.18 It is through investigations such as this that novel candidate markers are being identified.
RNA-based markers include genes that are overexpressed or underexpressed as transcripts of regulatory RNAs (microRNAs).19 Differentially expressed genes in a particular tumor are detectable for thoracic malignancies such as NSCLC or esophageal cancer in the lymph nodes and serum of patients. No one single marker has proved ideal as a predictor of cancer. Recent efforts have focused on a panel of markers for most thoracic malignancies. Patterns of markers can more accurately predict cancer than a single molecule can, but choosing which genes to include in an analysis requires a multidisciplinary integration of biostatistics and bioinformatics. We await the results of large prospective studies evaluating various markers. MicroRNAs (miRNAs) are an abundant class of small non-protein-coding RNAs that function as negative gene regulators in the cell. Investigations have suggested that a single miRNA could bind to a number of mRNA targets; these targets could be implicated in cellular transformation and oncogenesis. This area of investigation is new, but it has already been shown that miRNA expression profiles can be used to classify human cancers.20 In lung cancer, miRNA expression profiles and specific miRNAs have been shown to correlate with prognosis and survival.19,21 The emerging findings on miRNAs are very exciting; further development is needed, along with integration with other molecular marker technology.
MICROARRAY ANALYSIS OF PRIMARY TUMORS
Briefly, a DNA microarray comprises multiple, gene-specific polynucleotides (probes) arranged along exact coordinates of a two-dimensional grid (array), individually immobilized to a single substrate.22 Total RNA pools from experimental or reference specimens are reverse-transcribed to fluorescence-labeled complementary DNA (cDNA) before incubation with the microarray (Fig. 45-1A). This process allows simultaneous quantitation of the relative (experimental versus reference) amount of messenger RNA (mRNA) transcripts. The number (which may be hundreds of thousands) of immobilized microarray probes determines the amount of gene expression information returned from each microarray experiment. This method depends on the highly sensitive and specific hybridization between complementary strands of nucleic acids.
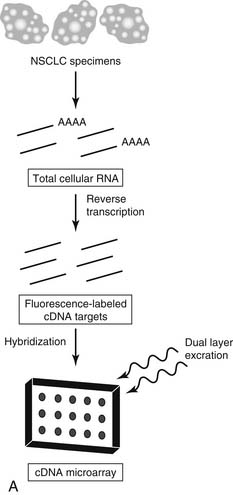
Microarray platforms are of two general types: cDNA and oligonucleotide. Typically, the cDNA microarray contains hundreds to tens of thousands of long, polymerase chain reaction–derived representations of specific genes (about 600 to 2400 base pairs). In contrast, the oligonucleotide microarray contains tens of thousands to hundreds of thousands of 25-mer oligonucleotides complementary to unique gene sequences, directly deposited by light-directed chemical synthesis.23 Currently, no consensus exists as to the ideal type of microarray platform. Each has distinct advantages and drawbacks that are beyond the scope of this discussion. In general, the data sets derived from each platform are identical; each allows for large-scale analysis of gene expression in biologic specimens from a single experiment. Functionally, most investigators have gravitated toward the Affymetrix-type chip.
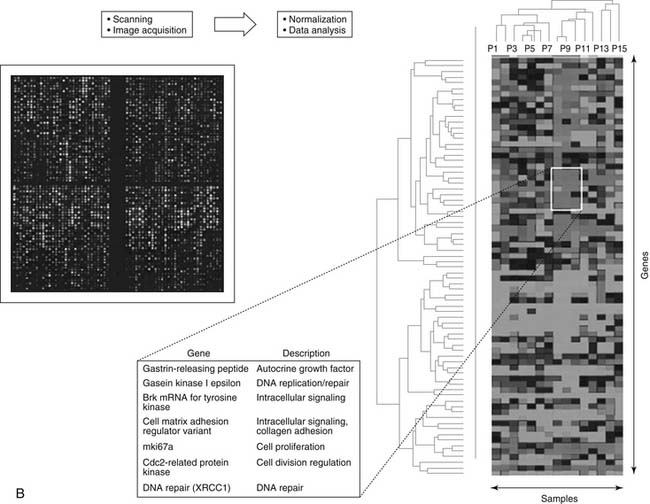
The vast amounts of data generated by microarray analyses have necessitated evolution in bioinformatics—a field concerned with the acquisition, storage, display, and analysis of genome-wide expression data. So many options are now available for analysis or data mining that choosing among them is challenging. The predominant analytic method in current use is a family of algorithms referred to as cluster analysis. For example, one specific type of algorithm is hierarchical clustering. The term cluster analysis generally applies to methods for organizing multivariate data into groups with similar patterns, so that they may impart additional information about certain genes and the study specimens (see Fig. 45-1B).24 Overall, data analysis can be divided into four common themes: detection of differential gene expression, pattern discovery, prediction of specimen characteristics, and inference of molecular pathways and networks. The existing microarray literature on lung and esophageal cancers has addressed the first three of those four themes of analysis and has reported analyses and hypotheses regarding the molecular pathways and networks of importance.
Initial NSCLC microarray profiling studies were conducted either on cell culture systems25,26 or on matched tumor versus nontumor specimens,27,28 to identify genes with significantly altered expression levels. Many of the genes differentially expressed in tumor specimens were involved in cell cycle regulation, cell metabolism and signaling, or apoptosis; the findings of those initial NSCLC microarray profiling studies were consistent with the findings of studies in the broader cancer literature. However, some genes were identified that were not commonly associated with NSCLC: they suggest previously unrecognized mechanisms in tumorigenesis, or perhaps they represent novel therapeutic targets (or both).
The most interesting recent investigations have focused on patients with early-stage NSCLC. This focus has been clearly advantageous because including patients with more advanced disease adds many clinical variables that complicate their prognosis. Several groups have been able to identify gene expression profiles between 22 and 50 genes that can separate otherwise identical tumors in terms of disease-free survival and overall survival rates after potentially curative resection.29–32 Bhattacharjee and colleagues studied 186 NSCLC tumors using a clustering technique specialized for gene pattern discovery; they identified four distinct, novel subgroups of adenocarcinomas according to gene expression profiles30 and concluded that lung cancer diagnosis could be improved by integrating expression profile data.
Beer and colleagues constructed a risk index based on a set of 50 genes identified by hierarchical clustering and univariate Cox analysis that could predict survival in early-stage lung adenocarcinoma patients.29 Members of the gene set included genes not previously associated with survival, suggesting novel therapeutic targets. They stated that a gene set predictive of survival in early-stage adenocarcinoma might identify a high-risk subgroup who could benefit from adjuvant therapy.
Kikuchi and coworkers microdissected individual cancer cells from 37 NSCLC tumors before performing microarray analysis to identify novel tumorigenesis genes as well as genes related to chemosensitivity.33 Their subset cluster analysis of 18 specimens identified 40 genes whose expression levels could discriminate gross, pathologically confirmed lymph node metastasis.33 They speculated that such gene profile–based characteristics of NSCLC tumors could eventually guide personalized therapies for patients.
Regardless of the study, larger-scale prospective studies are required. Some overlap occurred between profiles in the studies described here, but there were also some distinct differences that may be attributed to varying microarray technique, samples, and statistical methods. To resolve some of these issues, the National Cancer Institute issued a Director’s Challenge recently, which led to the first dataset whose main aim was to establish uniformity across different subject populations and different laboratories. Through a highly coordinated effort, the authors recently reported their results of their large, training-testing, multisite, blinded validation study. They were able to characterize the performance of several prognostic models by reviewing the gene expression profiles for 442 lung adenocarcinomas.34 Their models performed best when microarray data were combined with basic clinical variables of tumor stage, patient age, and patient sex. This was able to predict overall survival in the study population. Furthermore, their results support the combined use of clinical and molecular information in the prognostic modeling of early-stage NSCLC.
Microarray studies of esophageal tumors have been generally small in terms of the number of tumors analyzed, but some early insights have been noteworthy. Selaru and colleagues used hierarchical cluster analysis in an attempt to classify esophageal tumor specimens by gene expression profiles. As predicted, they found that specimens from patients with Barrett’s esophagus and from patients with esophageal cancer clustered separately.35 In addition, they were able to accurately subgroup squamous cell carcinomas and adenocarcinomas by comparing cluster analysis findings with histopathologic subtypes.
Other investigators have used microarray analysis to gain insight into the progression of esophageal tumors from dysplasia to metastasis. Using a cancer gene–specific microarray of 588 genes, Zhou and coworkers identified sets of differentially expressed genes in normal esophageal mucosa, basal cell hyperplasia, high-grade dysplasia, carcinoma in situ, and overt cancer.36 Their analysis showed that two genes (P160ROCK and JNK2) in particular may play an important role in carcinogenesis. Their results are potentially powerful, because they evaluated gene expression across different stages of carcinogenesis. In their analysis, the P160ROCK gene (a member of the family of Rho-associated serine/threonine kinase isoenzymes that regulate cell motility and morphologic changes) was dramatically upregulated in the tissues with high-grade dysplasia. Thus, its activation may be one of the early events in carcinogenesis of esophageal tumors.
Further, Wang and colleagues used transcriptional profiling with microarrays of esophageal tissues at various stages of carcinogenesis from patients with Barrett’s esophagus and esophageal adenocarcinoma.37 They identified 36 genes that were differentially modulated according to microarray data in Barrett’s neoplastic progression; 12 genes were significantly differentially expressed in cancer-associated Barrett’s, a gene set that they hypothesized represents a biomarker profile for adenocarcinoma at early stages.
Additionally, Kihara and coworkers analyzed the expression profiles of 20 surgically resected esophageal tumors in patients treated with adjuvant therapy.38 They identified a profile of 52 genes that were correlated with patient’s prognosis, and they found that the gene set accurately predicted the chemosensitivity and chemoresistance of the tumors to various agents. From this gene set, they obtained a “drug response score” that correlated significantly with prognosis.
More recently, others have performed similar analyses in patients with squamous cell carcinoma of the esophagus, with the goal of identifying novel markers for prognosis and treatment.39 The investigation of esophageal cancer has substantially progressed in the past few years, but it remains hampered by the lack of studies with large numbers of specimens and the lack of a unified commitment to standardization (as compared with the large studies of NSCLC). Prospective verification of genes and predictive models is also needed in studies of esophageal cancer.
Mesothelioma is another malignancy that has been the target of gene expression analysis, although relatively few studies have evaluated it to date, in part because of its rare incidence and because of the difficulty in acquiring suitable tissue for analysis. Most reported studies have been performed on malignant pleural mesothelioma (MPM) cell lines and have provided only descriptive data. Our group at the University of Minnesota has attempted to make some sense of this by incorporating primary tumor specimens into the array analysis with hierarchical clustering.40 In our analysis, we identified several genes associated with MPM and also noted matriptase overexpression (826-fold)—clues to the mechanisms of oncogenic transformation. Not surprisingly, other investigators have performed similar studies with alternative findings, which highlights the inherent variability of the approach used. Still other groups have expanded the analysis of primary tumor specimens. Gordon comprehensively reviewed the mesothelioma studies,41 and Gordon and coworkers highlighted the recent attempts at translating microarray data into clinically relevant prognostic information.42,43 They used a gene expression ratio–based prognostic analysis of MPM tissues in an attempt to predict and validate the clinical outcome in 39 independent MPM tumor specimens. They theorized that using such gene ratios to translate gene expression data into easily reproducible, statistically validated clinical tests will help facilitate clinical decision making in the future.
MOLECULAR DETECTION OF OCCULT MICROMETASTASES
The detection of regional thoracic lymph node metastases by routine histopathologic analysis portends a significantly diminished patient survival rate, usually secondary to the later development of systemic metastases in NSCLC. However, even with N0 status, the 5-year survival rate of patients with T1 and T2 tumors combined is about 60% to 70%, indicating that the undetected presence of occult micrometastases (OM) in either lymph nodes or other systemic sites might be clinically significant. The ability to accurately detect OM would allow better prediction of the risk of tumor recurrence and of death, as well as the use of targeted adjuvant therapy in high-risk patients. The term occult micrometastases has been criticized by some as ambiguous, but it is now commonly used, even though it has been suggested that a better term would be occult tumor cells.44 In this chapter, we will use OM.
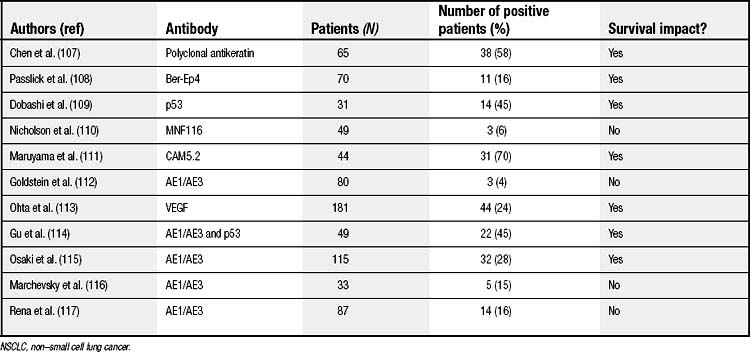
Two techniques are primarily used to detect OM: immunohistochemistry (IHC) (Fig. 45-2) and polymerase chain reaction (PCR) (Fig. 45-3). With respect to IHC, several groups have investigated the utility of antibodies (Abs) to various tumor markers in detecting OM in the lymph nodes of patients with surgically resected stage I NSCLC. Such Abs include, for example, anti-cytokeratin monoclonal Abs (mAbs) (MNF116, CAM5.2, and AE1/AE3), Ber-Ep4 (an epithelial cell–specific mAb), and anti-p53 mAb. Detection rates have varied widely, with a change in nodal status occurring in 4% to 58% of lymph nodes. Of the six largest studies, four showed a direct correlation between positive IHC results and an increased risk of cancer relapse (Table 45-1). Two clinical trials, the Cancer and Leukemia Group B (CALGB) 9761 and the American College of Surgeons Oncology Group, are in the process of defining, in a prospective, multi-institutional manner, the relationship between lymph node OM detected by IHC and systemic tumor recurrence. Preliminary analysis of CALGB 9761 IHC results demonstrated an upstaging of 11% of the 178 patients.45 Final analysis of the results of the CALGB 9761 trial is currently being performed.
The detection of individual tumor cells in patients’ bone marrow is another area of active interest. Passlick’s group has demonstrated that IHC is a valid and reliable approach for detecting occult disseminated tumor cells in bone marrow.46 Using cytokeratin IHC, several groups have demonstrated a 22% to 59% incidence of bone marrow micrometastases in NSCLC.46–49 An increasing number of published articles have elucidated the potential prognostic importance of this finding.46,47,49,50
In the past 10 years, a number of studies of several different malignancies have used reverse transcriptase-PCR (RT-PCR) to detect micrometastatic disease, including thoracic tumors such as NSCLC. RT-PCR detects micrometastatic disease by identifying an appropriate tumor marker. A number of molecular markers have been investigated in patients with NSCLC. Apo-mucin type 1 (MUC1) mRNA, a mucopolysaccharide gene associated with epithelial tissues, was the first marker used in NSCLC lymph nodes to identify micrometastatic disease in patients with surgically resected NSCLC.51 Subsequent studies have revealed the ubiquitousness of MUC1 mRNA in many tissues, diminishing its value as a tumor marker52–54 (and Maddaus and D’Cunha, unpublished data). Other markers need to be identified in patients with NSCLC.
Carcinoembryonic antigen (CEA) has emerged as a potentially suitable NSCLC tumor marker from a number of RT-PCR–based studies.55–58 Collectively, those studies demonstrated that CEA has relatively high specificity (mRNA transcripts were detectable in almost all epithelial cells, including cancer cells, but not in nonepithelial cells) and a relatively high sensitivity (it detected one malignant cell in up to 106 normal cells). Our group, within the CALGB 9761 trial, used RT-PCR to examine the potential of CEA as a marker of OM in lymph nodes of patients with early-stage NSCLC; our results definitively expanded on previous findings.59 Standard and quantitative real-time RT-PCR for CEA detected OM in patients with stage I NSCLC at similar rates, with both methods upstaging patients in about 50% of cases.
A potential shortcoming of OM detection by both IHC and RT-PCR is the binary (yes-or-no) result regarding the presence or absence of OM. Although predictive of an increased risk of tumor recurrence, simple yes-or-no results fail to further stratify risk. Studies of N2 disease detected by hematoxylin and eosin (H&E) staining revealed significant survival differences between patients with lymph nodes replaced by tumors and patients with microscopic disease found incidentally after surgical resection.60 Similarly, rates of tumor recurrence and of survival in patients with lymph nodes negative by H&E although they harbor OM may also be related to the number of micrometastatic tumor cells present.
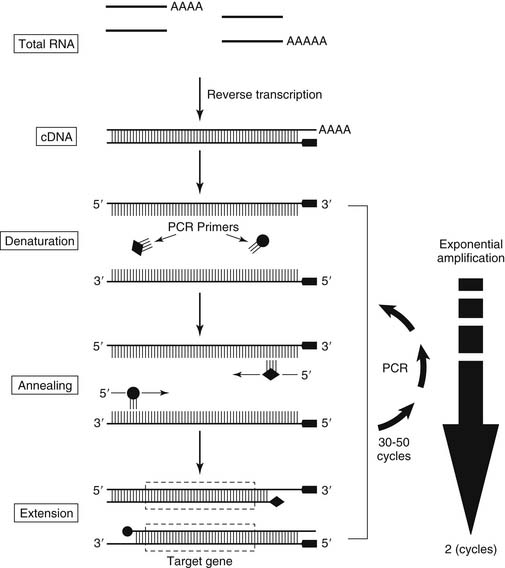
A possible solution may be the use of quantitative real-time RT-PCR (QRT-PCR), a technology now broadly available.61 QRT-PCR, unlike other approaches to quantifying PCR products, requires no post-PCR product manipulation (such as gel electrophoresis). Moreover, QRT-PCR enables rapid processing of many samples (96 reactions in 3 to 4 hours). QRT-PCR uses a target molecule–specific oligonucleotide probe (in this case to CEA mRNA) with a covalently attached fluorescent reporter and quencher dye. When the probe is intact, the quenching dye absorbs the fluorescent energy of the reporter dye. If the target molecule of interest is present, the specific oligonucleotide probe anneals to it. During the PCR extension phase, the fluorescent probe is cleaved by the exonuclease activity of Taq DNA polymerase, thus increasing the reporter dye’s emission. Curves plotting the relative fluorescence change against the PCR cycle number are constructed, and the point of increase in fluorescence above background is calculated; from this calculation, the threshold cycle (CT) is determined. Because CT values decrease linearly with increasing target molecule quantity in the input sample, they provide a quantitative measurement.
The potential application of QRT-PCR was demonstrated in the same group of 53 patients in our CALGB 9761 study cited previously: of 232 lymph nodes, 59 (25.4%) were positive by QRT-PCR for CEA mRNA. Estimates of the quantity of micrometastatic cells in those 59 nodes ranged from 1.1 × 103 to 3.2 × 105 cells per lymph node station (median, 7190 tumor cells per lymph node station). The increased sensitivity of this molecular approach (versus the standard H&E analysis) accounted for the additional seven patients whose tumors were upstaged. Others have subsequently confirmed our findings and suggested a prognostic relationship.62,63 The prognostic importance of OM in NSCLC awaits the conclusion of the CALGB 9761 (manuscript in preparation).
Numerous other markers have been investigated in NSCLC by QRT-PCR, including cytokeratin 7, cytokeratin 19, KS1/4, LUNX, and PDEF. This list of markers is not meant to be all-inclusive but rather to highlight the fact that many markers exist. The single-institution studies of such markers have looked at limited numbers of patients, yet the vast majority of those reports suggested some prognostic significance.64,65 KS1/4 is an intriguing marker because it has a very high specificity for lung cancer.66 KS1/4 is a gene that encodes a glycoprotein expressed in epithelial cells recognized by the monoclonal antibody Ber-EP4. Monoclonal antibodies to Ber-EP4 (such as edrecolomab) have been used in IHC studies of NSCLC lymph nodes to identify micrometastatic disease. Furthermore, antibodies to Ber-EP4 have shown predictive promise in clinical trials for patients with colorectal cancer. Using edrecolomab, 189 patients with resected stage III colorectal cancer had a 32% increase in overall survival rate, as compared with the no-treatment arm (P < .01), and a decreased tumor recurrence rate (P < .04).67 Those results raise both staging and therapeutic issues.
An analogous approach has been used to evaluate the lymph nodes of patients with esophageal cancer. In esophageal cancer, lymph node involvement is the strongest predictor of recurrence and poor outcome. As with studies evaluating the prognostic importance of IHC-detected NSCLC micrometastases, the prognostic importance of IHC-detected lymph node involvement in patients with esophageal cancer has varied.68–72 This variation underscores one of the potential pitfalls of IHC—namely, sampling error from evaluating only a limited number of sections of the lymph node.
Luketich’s group (Godfrey and colleagues) used QRT-PCR to detect micrometastases in 387 lymph nodes from 30 histologically node-negative patients.73 In their report focusing on primary adenocarcinomas, CEA expression was used to identify 11 additional patients who had micrometastases. Of those 11 patients, 9 suffered disease recurrence. The Kaplan-Meier analysis demonstrated that QRT-PCR–positive lymph nodes correlated with a significantly lower disease-free survival rate (P < .0001) and a significantly lower overall survival (P < .0006). Other similar studies have followed, including one that evaluated over 39 different markers in esophageal adenocarcinoma from Luketich’s group.74 Finally, Inoue and colleagues used CEA RT-PCR in 57 patients with esophageal carcinoma (primarily squamous cell, histologically) to show that CEA-positive cells could be detected in 70% of patients.75 Their study lacked prognostic significance, however, as did a similar study with 48 patients who had squamous cell histology.76



