Fig. 22.1
CPFE syndrome at chest CT in a patient with rheumatoid arthritis (male, smoker). (a) upper lobes showing centrilobular and paraseptal emphysema; (b) mid regions of the lungs, showing predominantly paraseptal emphysema, with thickening of the interlobular septa; (c) lower zones showing usual interstitial pneumonia pattern with reticulation, honeycombing, and traction bronchiectasis
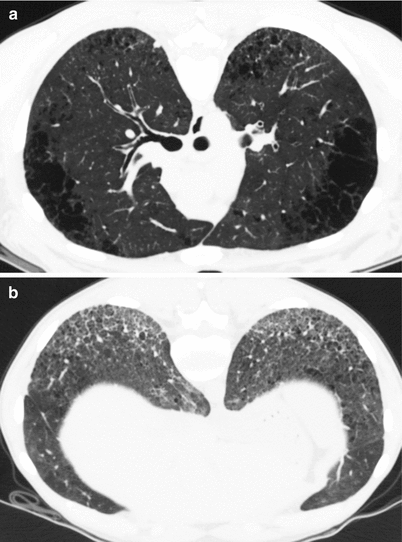
Fig. 22.2
CPFE syndrome at chest CT in a 28-year old female patient with severe systemic sclerosis and high-titer anti-U1-RNP autoantibodies, with a smoking history of less than 5 pack-years. (a) Upper lobes demonstrating centrilobular and paraseptal emphysema; (b) lower zones showing nonspecific interstitial pneumonia pattern with reticulation, ground-glass opacities, and mild traction bronchiectasis
In 150 consecutive patients with rheumatoid arthritis [45], 19 % had ILD, 15 % had so-called “emphysematous bullae”, including 8 % (12 out of 150) who had both ILD and emphysema. Emphysema was observed in 24 % of 63 patients with rheumatoid arthritis and ILD [46], and was significantly more frequent among patients with a CT pattern of UIP (38 %) than in other ILD patterns; patients with UIP also had a higher prevalence of smoking and a greater smoking history than those with other patterns. Antoniou et al. reported the presence of emphysema at chest CT in 48 % of ever-smoker patients with rheumatoid arthritis and ILD, and in 35 % of ever-smoker patients with IPF, despite median smoking histories of less than 25 pack-years in both cohorts [47]. These data suggest that subjects with rheumatoid arthritis who smoke may be particularly vulnerable to emphysema. In addition, patients with rheumatoid arthritis and a history of smoking had a “coarser” fibrosis at imaging than never smokers. In rheumatoid arthritis, interaction of genetic background (the human leukocyte antigen DRB1 shared epitope) with environmental exposures (especially tobacco smoking) and autoimmunity (anti-cyclic citrullinated peptide antibodies) is now well established [48]. Briefly, tobacco smoking is responsible for inflammation and citrullination of proteins within the lung, a post-translational modification that alters L-arginine residues into L-citrulline residues through peptidyl arginine deiminase, thereby altering protein folding and charge, with enhanced degradation by proteases, and exposure of cryptic epitopes, which in turn increases the risk of developing anti-cyclic citrullinated peptide auto-antibodies produced in the lung, and then rheumatoid arthritis [49]. In this process, the immune response developed against modified self-proteins transfers inflammation from the lungs to the joints [50]. Tobacco smoking increases the incidence and severity of rheumatoid arthritis [51], with furthermore a higher risk of developing extra-articular complications [52] possibly including ILD [53, 54]. The ILD may exacerbate in patients with CPFE and rheumatoid arthritis, occasionally upon drug therapy [55]. Whether emphysema in rheumatoid arthritis is also a consequence of autoimmune processes promoted by cigarette smoking is unknown. Of note, anti-elastin antibodies are not found in patients with CPFE [56].
CT evidence of emphysema was found in 8.4 % of 225 patients with systemic sclerosis [57], with extent of emphysema greater than 10 % in about a quarter of these, as compared to 12 % of patients with IPF and 22 % of patients with idiopathic NSIP. Tobacco smoking negatively influences FEV1:FVC and DLco in patients with systemic sclerosis [58]. Emphysema is more prevalent in systemic sclerosis patients with pulmonary fibrosis than in control smokers with connective tissue disease or IPF, after adjustment for the smoking history [59]. The CPFE syndrome may occur in patients with only mild smoking history [60, 61], suggesting that systemic sclerosis itself might contribute to the development of emphysema, a hypothesis that remains to be explored. Spontaneous emphysema and right heart hypertrophy develop spontaneously in tight-skin mice that harbor a duplication in the fibrillin–1 gene and resemble human systemic sclerosis [62, 63]. Modulation of inflammatory markers in animal models [64–69] supports the hypothesis that the connective tissue disease per se may play a role in the pathogenesis of the CPFE syndrome, possibly through chronic inflammation or epigenetic dysregulation (post-translational modifications of histone proteins and hypermethylation) [70, 71].
CPFE has been reported in patients with polymyositis, Sjögren’s syndrome, mixed connective tissue disease, overlapping connective tissue disease, with consistent antibody profiles [21]. Emphysematous changes were found in 3 of 65 autopsy cases of polymyositis/dermatomyositis [72]. Tobacco smoking also increases the risk of developing systemic lupus erythematosus [73], however CPFE seems rare in lupus.
The CPFE syndrome may occur in patients with so-called interstitial pneumonitis with autoimmune features (also referred to as ILD in undifferentiated connective tissue disease [74], auto-immune featured connective tissue disease [75] or lung-dominant connective tissue disease [76]), e.g. the recently individualised condition with manifestations suggestive of connective tissue disease but not satisfying the criteria of any defined disease entity [77]. For example, a syndrome of CPFE was found in 7 % of subjects with lung disease and anti-cyclic citrullinated peptide antibodies but not rheumatoid arthritis [78].
The CPFE syndrome may also develop in patients with systemic vasculitis especially microscopic polyangiitis [79]. In one study, autoimmune markers including perinuclear anti-neutrophil cytoplasmic antibodies were more frequently found in patients with CPFE than in subjects with IPF and no emphysema, correlating with infiltration of the fibrotic lungs by clusters of CD20+ B lymphocytes within lymphoid follicles [80]. It is speculated that anti-myeloperoxidase antibodies in microscopic polyangiitis may promote the degranulation of neutrophils, with release of reactive oxygen species that may participate to disease pathogenesis.
Other Etiological Contexts
CPFE has been occasionally reported in subjects with exposure to agrochemical compounds [81], coal dust [82, 83], talc [84], or to rare earth elements and tobacco [85]. In addition, CPFE was reported in patients with asbestosis [12, 13, 86, 87], silicosis [88], and sarcoidosis [89], occasionally in the absence of tobacco smoking. Subjects exposed to mineral dust and especially coal dust may develop emphysema and pulmonary fibrosis simultaneously [19]. The combination of emphysema and ILD has been further reported in the setting of farmer’s lung [90, 91].
Clinical Manifestations
Patients with the CPFE syndrome have a mean age of 65–70 years [7, 19], with younger individuals especially in those with connective tissue disease or genetic predisposition to ILD (See Clinical Vignette). The male:female ratio is greater than 9:1 in CPFE, with 60 men and only 1 woman in the seminal series [7]. In patients with connective tissue disease, the CPFE syndrome is less strongly associated with male gender (68 % of males), however male predominance in patients with CPFE does contrast with the female predominance found in series of patients with connective disease and ILD (without emphysema) [92].
Patients with CPFE usually report severe dyspnea at exercise [2, 6, 7, 14, 17, 18, 20, 21, 81, 93–95]. Chronic bronchitis may be present. Clinical examination generally demonstrates basal “velcro” crackles similar to that found in IPF. Finger clubbing is present in one-third of the patients [21].
Clinical Vignette
A 68-year old male, ex-smoker, with a history of 48 pack-years, who had worked as a mason, with no significant exposure to asbestosis, was referred to the pulmonary clinic for severe dyspnea on exertion. He had a history of coronary heart disease, with myocardial infarction and coronary stenting 4 years prior to admission. Velcro-crackle rales of the lung bases were present at lung auscultation. The chest radiograph demonstrated mild hyperlucency of the upper zones, with hyperinflation, and reticular changes of the lower zones. Chest CT demonstrated both emphysema of the upper zones (centrilobular and paraseptal) and fibrosis of the lung bases, with subpleural reticulation, honeycombing, and traction bronchiectasis, with non-prominent superimposed ground glass attenuation (Fig. 22.5). Areas of admixed pulmonary fibrosis and emphysema were observed in the mid sections of the lungs. Pulmonary function tests were: FVC 86 % of predicted value, FEV1 78 % of predicted, FEV1:FVC 0.68, TLC 89 %, RV 117 %, DLco 44 %, Kco 57 %, PaO2 at rest 65 mmHg. Echocardiography showed slightly dilated right heart cavities with estimated systolic pulmonary artery pressure of 42 mmHg. No clinical signs of connective tissue disease were present, and antinuclear antibodies were negative. The patient was diagnosed with combined pulmonary fibrosis and emphysema syndrome at multidisciplinary discussion, with possible UIP pattern at CT. Lung biopsy was not performed. Inhaled bronchodilators were initiated. Fourteen months later, he was readmitted for acute right heart failure. Right heart catheterisation demonstrated severe precapillary pulmonary hypertension, with mean pulmonary artery pressure of 42 mmHg, pulmonary artery wedge pressure of 12 mmHg, and cardiac index of 1.9 L/min/m2. Sildenafil was initiated in the setting of a prospective registry, with moderate hemodynamic improvement at 3 months, and unclear clinical benefit. The patient died 5 months after the diagnosis of pulmonary hypertension from acute respiratory failure.
Pulmonary Function and Physiology
Patients with CPFE syndrome present with limitation to exercise capacity, and severely impaired DLco and transfer coefficient (Kco), contrasting with subnormal spirometry [19, 21]. Spirometric values and lung volumes are preserved, or with FVC, TLC, and/or FEV1:FVC close to the lower limit of normal. In our series, the mean FVC was 90 ± 18 % of predicted value, TLC was 88 ± 17 %, FEV1 was 80 ± 21 %, FEV1:FVC was 89 ± 13 %, whereas DLco was 37 ± 16 % of predicted and Kco was 46 ± 19 %. In patients with CPFE and associated PH [20], lung volumes were comparable with FVC of 88 ± 18 % of prediced, however DLco was only 24 ± 14 % and Kco was 28 ± 16 % of predicted.
Merged data from three study populations [7, 20, 21] indicated that only 36 % of 132 patients had TLC lower than 80 % of predicted values. Only 41 % of 132 patients had FEV1:FVC lower than 0.70 (out of whom 11 % had FEV1 greater than 80 % of predicted, corresponding to GOLD [Global initiative for Obstructive Lung Disease] stage 1); 37 % were classified as GOLD 2009 stage 0 (FEV1:FVC ≥0.70 and FEV1 ≥80 % of predicted) and further 22 % were unclassified according to GOLD 2009 (with FEV1:FVC ≥0.70 and FEV1 <80 % of predicted). In another study, smokers with emphysema were less likely to meet GOLD criteria for chronic obstructive pulmonary disease if ILD changes were present at imaging [96]. Thus, the relative preservation of spirometric values may lead to underdiagnosis of the CPFE syndrome.
These observations are attributed to the counterbalancing effects of the restrictive physiology associated to the elastic forces increased by pulmonary fibrosis (with presumably increased elastic recoil, as well as prevention by traction forces of expiratory airway collapse), and the effects of the obstructive physiology with propensity to hyperinflation due to emphysema. This is illustrated by the possibility of FEV1:FVC to actually improve back to normal values while the disease progresses, with worsening of dyspnea and DLco [97]. The annual change in FEV1:FVC has been shown to moderately increase in patients with CPFE, as compared to a more profound annual decrease in those with chronic obstructive pulmonary disease [98]. TLC correlates positively with the emphysema score at HRCT, and inversely correlates with the fibrosis score; conversely, FEV1:FVC negatively correlates with the emphysema score at HRCT, and positively correlates with FEV1:FVC [99]. Analysis of respiratory impedance by multi-frequency forced oscillation technique found lower whole-breath, inspiratory or expiratory resistance in CPFE patients than in chronic obstructive pulmonary disease, and lower whole-breath and expiratory resistance in CPFE than in ILD without emphysema, further supporting the hypothesis of pseudonormalisation of lung mechanics in CPFE [100]. Conversely, both disease components concur to reduced alveolar capillar gas exchange through either decreased capillary blood volume or alveolar membrane thickening.
Severe decrease in arterial oxygen saturation and hypoxemia at exercise even of minor intensity is very common, especially when CPFE is complicated by severe PH. In our series of 61 patients [7], the room air partial pressure of oxygen in arterial blood (PaO2) decreased at exercise (20–50 W) by a mean of 1.5 ± 1.6 kPa (11.2 ± 12 mmHg). During a 6-min walk distance test, the arterial oxygen saturation measured by pulse oxymetry decreased by 9 ± 6 %. In another group of patients with CPFE and PH, the arterial oxygen saturation measured by pulse oxymetry decreased by 15 ± 8 % [20]. Hence, exercise limitation with decrease in oxygen saturation, and isolated [101] and/or severe [102] reduction in DLco or Kco contrasting with mild ventilatory defect or normal spirometry should raise the suspicion for CPFE syndrome. Hypercarbia occurs only very late in the disease course and patients may die from the physiological consequences of hypoxia before significant hypercarbia takes place.
As compared to patients with IPF and no emphysema, those with CPFE have higher lung volumes (FVC and TLC), generally comparable FEV1 and residual volume (RV), lower DLco, and lower PaO2 [3, 18, 103, 104]. The mean FEV1:FVC is within the normal range or close to the lower limit of normal in CPFE, however it is lower than in IPF where it is usually increased (e.g. greater than 0.80) [103, 104]. Comparison of physiology between groups may be hampered by differences between studies in the severity of emphysema, fibrosis, and emphysema versus fibrosis, despite attempts to adjust for severity of fibrosis [18]. Demographics of CPFE and IPF are similar in those studies, however patients with CPFE tend to have greater tobacco smoking history [18, 103]. As expected, FEV1 and FEV1:FVC are preserved in patients with CPFE as compared to those with chronic obstructive pulmonary disease, who also tend to have more hyperinflation and less altered transfer capacity [98].
Importantly, the presence of significant emphysema impacts longitudinal lung volume measurement, attenuating the effect of fibrosis on lung function parameters. Patients with CPFE experience a slower decline in FVC and DLco than IPF patients without emphysema [103, 105]. Therefore, changes in FVC and DLco are not reliable indicators of disease progression in patients with CPFE. As most recent clinical trials in IPF use FVC as an endpoint, it is preferable that patients with CPFE be excluded from IPF trials [11], and similarly from trials of ILD in connective tissue disease [42]. In clinical practice, serial changes in FVC and DLco are used to monitor disease progression in IPF [27], but they are not appropriate in CPFE patients, with unfortunately no clear-cut alternate functional parameter proposed so far. In one study, a decline in FEV1 by 10 % or more at 6 or 12 months was useful to assess disease progression, and predicted a poor outcome [17]; FEV1:FVC might also be useful [106]. Whether these observations are useful in the clinic awaits further evaluation.
Imaging
Chest radiograph may show hyperlucency of the upper zones of the lungs, and diffuse parenchymal infiltrates in the lower lobes (Fig. 22.3). HRCT of the chest has dramatically enhanced the recognition of CPFE and is key to the diagnosis. Patients with CPFE present with both emphysema (generally predominating in the upper lobes) and features suggestive of pulmonary fibrosis (mostly in the lower lobes), with occasionally lung pathology available.
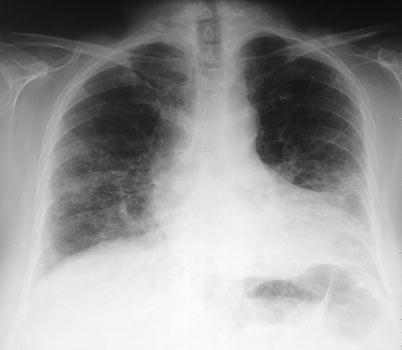
Fig. 22.3
Chest radiograph of CPFE syndrome showing hyperlucency of the upper zones of the lungs, and diffuse parenchymal infiltrates in the lower lobes (male, smoker)
Computed Tomography Characteristics
In our series [7], interstitial changes were characterized by honeycombing (95 %), reticulation (87 %), traction bronchiectasis (69 %), and architectural or bronchial distortion (39 %), predominating in the lower lung zones and in subpleural areas. Non-prominent ground-glass attenuation was present in two-thirds of the patients. Some degree of centrilobular emphysema was present in 97 % of the patients [7, 81]. In addition, paraseptal emphysema present in 93 % of patients with CPFE represented the predominant type of emphysema in more than half of patients [7]. Importantly, paraseptal emphysema seems to be more frequent in CPFE than in chronic obstructive pulmonary disease [93], and may actually be considered a hallmark of the syndrome [11, 81]. Panlobular emphysema is seldom observed. Signs of PH may be present at HRCT.
Thick-Walled Large Cysts
One peculiar pattern observed in CPFE is that of thick-walled large cysts (or “air spaces with fibrotic walls”) of the lower zones of the lungs [21, 42]. Thick-walled large cysts are frequently observed in areas where reticulation is present. They are larger than 2.5 cm in diameter and delimitated by a wall at least one mm thick. They may be associated or not with typical honeycombing, from which they differ by the larger size of the cysts and their often being not clustered, whereas honeycombing corresponds to clustered, cystic air spaces, 3–10 mm and up to 2.5 cm in diameter [107]. They may be associated with more extensive emphysema at imaging [108]. Thick-walled large cysts likely result from the development of pulmonary fibrosis in the setting of emphysematous lung, with enlargement of the cysts due to retraction forces in fibrotic lung [99, 109]. Enlargement over time of thick-walled large cysts has been described [108]. We consider that they are one typical feature of the CPFE syndrome [21].
Imaging Phenotypes
Due to the high heterogeneity of imaging in patients with CPFE, attempts were made to identify distinct imaging phenotypes [99] including (1) emphysema in the upper zones and fibrosis in the lung bases, with no or little overlap of emphysema and fibrosis in between (separate processes) (Fig. 22.4); (2) progressive transition from emphysema lesions to fibrosis, with significant overlap or admixture in mid areas (Fig. 22.5); (3) conspicuous paraseptal emphysema with predominant subpleural bullae/cysts with thickened walls (Fig. 22.6). The pattern of thick-walled large cysts (Fig. 22.7), described more recently [21], may overlap with that of predominant paraseptal emphysema. In addition, a number of observations do not fit into one of the above categories [99]. When adjusting for severity of fibrosis, patients with a pattern of predominant paraseptal emphysema had higher (e.g. normal) FEV1:FVC and lower FVC and TLC values, with similar DLco, as compared to those with separate processes and progressive transition between emphysema and fibrosis [99]. However, whether these subgroups have physiological relevance, and whether the pattern of predominant paraseptal emphysema really is the most associated with preserved spirometry, as shown in patients with chronic obstructive pulmonary disease [110], warrant confirmation.
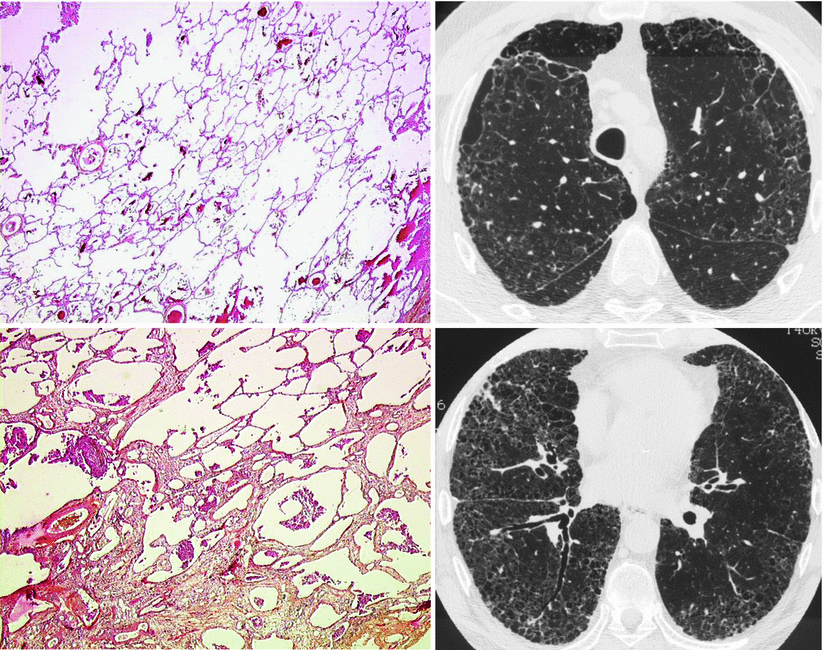
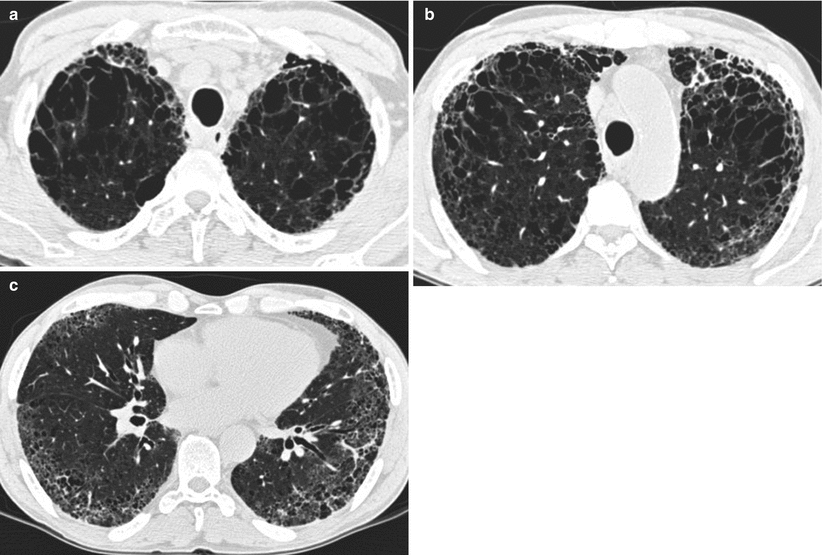
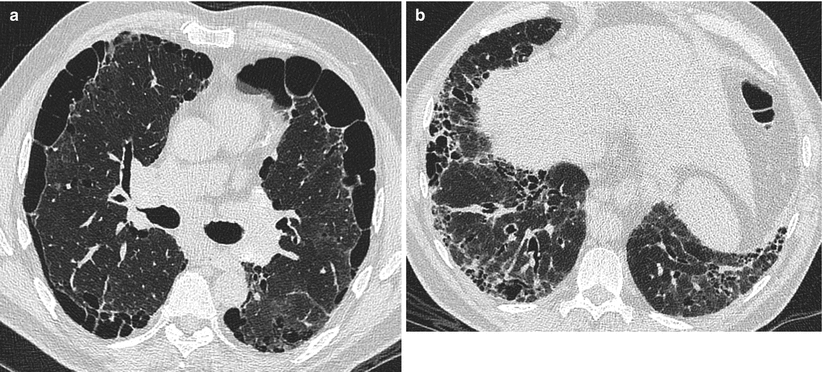
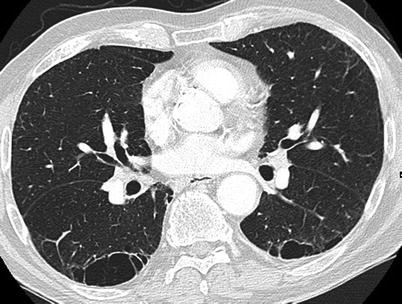
Fig. 22.4
CPFE syndrome with centrilobular emphysema and usual interstitial pneumonia (male, smoker). Upper left panel: lung biopsy in right upper lobe showing centrilobular emphysema; upper right panel: chest CT at the level of the trachea showing moderate centrilobular emphysema, along with reticular changes; lower left panel: lung biopsy in right lower lobe showing usual interstitial pneumonia pattern, with interstitial fibrosis and architectural distortion; lower right panel: chest CT in lower lung zones showing usual interstitial pneumonia pattern, with reticulation, traction bronchiectasis, honeycombing, and some ground-glass attenuation (Pathology slides courtesy of Dr Lara Chalabreysse, Lyon (France))
Fig. 22.5
CPFE syndrome at chest CT with centrilobular emphysema and usual interstitial pneumonia pattern, progressive transition phenotype (male, smoker). (a) Upper lobes showing centrilobular emphysema predominantly in anterior areas, with thickening of the interlobular septa; (b) mid regions of the lungs, showing admixture of centrilobular emphysema and fibrosis; (c) lower zones showing usual interstitial pneumonia pattern with reticulation, traction bronchiectasis, some honeycombing, and superimposed ground-glass attenuation predominating in the posterior areas
Fig. 22.6
CPFE syndrome at chest CT with predominantly paraseptal emphysema (male, smoker). (a) Mid Zones lobes showing predominantly paraseptal emphysema, with mild thickening of the interlobular septa; (b) lower zones showing fibrotic changes with honeycombing, traction bronchiectasis, and reticulation, predominating in the right lower lobe
Fig. 22.7
Chest CT showing thick-walled large cysts (male, ex-smoker). In this example, large subpleural cysts are relatively isolated, with little reticulation in the area surrounding the cysts. Septa are visible inside the right lower lobe cyst
Imaging Patterns
A majority of patients have a HRCT pattern of UIP (Figs. 22.1, 22.4, and 22.5) [7], however other patterns have been reported on imaging and/or histopathology [7, 111–114]. Similarly, mild to moderate ground glass attenuation is more prevalent in CPFE than in typical IPF patients [7], likely corresponding to NSIP (Fig. 22.2) [113] or to various smoking-related ILDs such as desquamative interstitial pneumonia [111] or respiratory bronchiolitis-associated ILD. Therefore, not all patients with (tobacco-related) CPFE have IPF [11], and not all patients with CPFE in the setting of connective tissue disease have a pattern of UIP [21].
Pitfalls
Interpretation of HRCT imaging is particularly difficult in patients with ILD and concurrent emphysema, owing to difficulties to ascertain honeycomb changes in patients with associated emphysema. In other words, association of a pattern of NSIP and emphysema [113], with small thick-walled cystic changes, may falsely resemble honeycombing at HRCT [114], a situation sometimes coined “possible honeycombing” [115]. Presence of emphysema is one of the main reasons for disagreement between radiologists in the CT assessment of honeycombing [116]. Because the recent international criteria for the diagnosis of IPF [117] are largely based on HRCT imaging and especially the presence of honeycombing (in patients without a lung biopsy), particular attention must be exerted to distinguish honeycombing from non-UIP interstitial changes with admixed emphysema, especially when emphysema is visible in the upper zones. Paraseptal emphysema is constituted by a single row of subpleural cystic spaces preferentially in the upper zones, contrasting to the clustered subpleural cysts (at least two rows) in honeycombing that predominates in the lung bases [118].
Diagnosis of the CPFE Syndrome
Although consensus diagnostic criteria have not yet been formally established, the CPFE syndrome is currently diagnosed on the presence of both emphysema (generally predominating in the upper lobes) and features suggestive of pulmonary fibrosis (mostly in the lower lobes on imaging), with occasionally lung pathology available.
However, it is unclear what extent of emphysema and fibrosis at imaging is needed to classify an individual has having CPFE rather than lone IPF (or emphysema with no significant ILD). The following diagnostic criteria used in the seminal description of the CPFE syndrome and later series [7, 20, 21] comprised: (1) “conspicuous” emphysema (centrilobular and/or paraseptal) defined as well-demarcated areas of low attenuation delimitated by a very thin wall (≤1 mm) or no wall; and (2) bibasilar reticular abnormalities with basal and subpleural predominance, traction bronchiectasis and/or honeycombing, and with minimal ground-glass opacities on HRCT scan. Although not based on semi-quantification of emphysema, this rather simple approach for diagnosing emphysema (e.g. noticeable without quantification of imaging features) has revealed to select patient populations with very reproducible physiology [7, 20, 21].
Other studies have defined CPFE as IPF (according to international criteria for the diagnosis of IPF at the time) with associated emphysema at imaging, using various thresholds or definitions for emphysema, including emphysema being notable or at least equivalent in extent to the fibrosis (“moderate emphysema”) [17]; or IPF with emphysema score ≥4/24 and fibrosis score ≥4/24, with scores based on estimates of the percentage of low attenuation areas (or of areas with reticulation/honeycombing) at three anatomic levels in both lungs (with emphysema and fibrosis considered significant when present in over half of the total lung fields) [100]; or IPF with >10 % of the lung affected with emphysematous changes [16]; or IPF with total emphysema score ≥10 % [18], a thresholds that corresponds to GOLD stage II or worse in patients with isolated chronic obstructive pulmonary disease; etc.
Provisional diagnostic criteria for the CPFE syndrome are listed in Table 22.1 [42]. The role of multidisciplinary discussion has not been formally studied in CPFE but is likely as important as in IPF (or more) [117]. Work in progress will likely refine diagnostic criteria for CPFE based on imaging as well as on lung physiology. Indeed, pulmonary function tests may actually contribute to the diagnosis of CPFE when demonstrating a typical functional profile, with preserved physiology and decreased DLco, especially in patients with mild imaging abnormalities. When present, thick-walled large cysts may contribute to the recognition of the syndrome. Similarly, precapillary PH in a patient with suspected CPFE may be considered as an additional clue to the diagnosis, as it is its most frequent complication present in about half of the patients. Conversely, CPFE may be diagnosed at an early stage as a result of screening for lung cancer [96, 119, 120], as described for IPF.
Table 22.1
Working diagnostic criteria for the syndrome of combined pulmonary fibrosis and emphysema (CPFE)
1. “Conspicuous” emphysema (centrilobular and/or paraseptal) at HRCT defined as well-demarcated areas of low attenuation delimitated by a very thin wall (≤1 mm) or no walla,b |
2. Bibasilar reticular abnormalities with basal and subpleural predominance, traction bronchiectasis and/or honeycombing on HRCT scanb,c |
3. Relatively preserved lung volumes and airflow with strongly decreased carbon monoxide transfer factord,e |
Biology
As in IPF and as observed in the general population, a minority of patients may have anti-nuclear antibodies, which in the absence of systemic clinical features have limited clinical relevance. As discussed above, anti-neutrophil cytoplasmic antibodies may also be found [80]. The serum levels of KL-6 (Klebs von der Lungen-6) and SP-D (surfactant protein-D) are elevated [121].
The differential cell count of bronchoalveolar lavage fluid is similar in CPFE to that of IPF and does not contribute significantly to the diagnosis [7, 71]; bronchoalveolar lavage is useful in the setting of acute worsening or suspicion of lung infection. Chemokines implicated in the recruitment of neutrophils (ENA-78/CXCL5 and IL-8/CXCL8) are elevated in the bronchoalveolar lavage fluid as compared to IPF [71] and may contribute to the development of emphysema.
Pathology
Due to emphysematous changes, frequent comorbidities, and severity of gas exchange impairment, very few patients with typical CPFE at imaging are subjected to lung biopsy by video-assisted thoracoscopic surgery. Therefore, only limited and likely skewed data are available regarding lung pathology in patients with CPFE. A variety of pathology patterns of ILD has been reported, including predominantly a UIP pattern (Fig. 22.8), NSIP, desquamative interstitial pneumonia (with extensive fibrosis), respiratory bronchiolitis – associated ILD [7, 19, 111–113, 122]. In an autopsy series of 22 cases including 15 (68 %) with a UIP pattern at HRCT – and 19 with lung cancer -, a pathological pattern of UIP was observed in all cases [108]. Honeycombing coexisting with emphysema was present in half of the patients. Interestingly, thick-walled large cysts at imaging corresponded pathologically to thick-walled cystic lesions lined by bronchiolar epithelium, located in the centriacinar/centrilobular region, involving one or more acini, with emphysematous changes and enlargement of membranous and respiratory bronchioles, dense collagen fibrosis of the walls, occasional fibroblastic foci, surrounded by honeycombing and normal alveoli. Thick-walled large cysts were not observed in IPF without emphysema [108]. Non-prominent bronchiolocentric fibrosis was occasionally present.
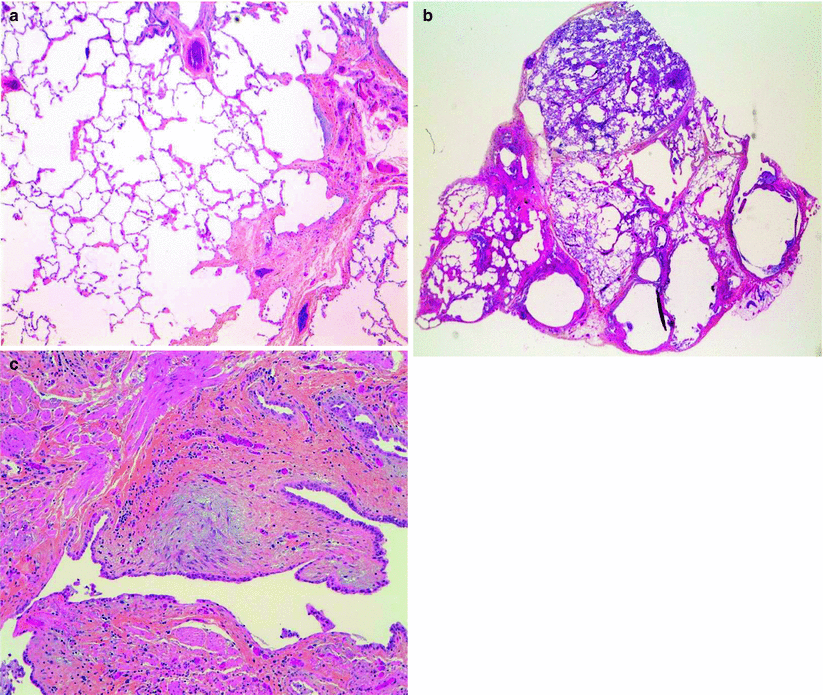
Fig. 22.8
Lung pathology in a patient with the CPFE syndrome. (a) Emphysema adjacent to interstitial fibrosis (upper right lobe); (b) dense fibrosis with subpleural pathological honeycombing (lower right lobe, low magnification); (c) fibroblastic focus in an area of dense fibrosis, at the center of the slide (lower right lobe, high magnification) (Courtesy of Pr Françoise Thivolet-Béjui, Lyon (France))
In addition, admixture of emphysematous changes and localized fibrosis with especially thickening of alveolar walls with little if any inflammation, and/or respiratory bronchiolitis, seems to be common in patients with CPFE. Histopathology of lung specimen in patients with emphysema has demonstrated thickened interstitium in addition to enlargement of alveolar spaces [123]. Excess of collagen and elastin deposition was present in the interstitium at electron microscopy, especially in septal walls of diseased areas [123]. Clinically occult interstitial fibrosis is surprisingly common in lobectomy specimens in smokers [124]. These abnormalities have been described with various terminologies with likely overlapping features [125]: “smoking related interstitial fibrosis” (Fig. 22.9) [126], “clinically occult interstitial fibrosis” [124], “airspace enlargement with fibrosis” [127], “fibrosis superimposed on emphysema” [125], “respiratory bronchiolitis-associated ILD” [128], “respiratory bronchiolitis-associated ILD with fibrosis” [122], and unclassifiable smoking-related interstitial fibrosis [124]. Such histopathological changes may only rarely be associated with physiological or radiological features of an ILD [125]. For the pathologist, the further presence of tobacco-laden alveolar macrophages is helpful as an indicator that the fibrosis is likely related to smoking and “superimposed on emphysema” [125].
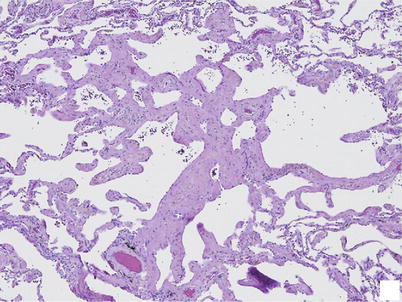
Fig. 22.9
Smoking related interstitial fibrosis. Severe interstitial fibrosis involving deep parenchyma in a centrilobular distribution associated with emphysema. Very little inflammation is present (From Katzenstein et al. [124], figure 2, panel B, with permission)
Collectively, these observations suggest that remodeling of the alveolar interstitium takes place in patients with predominant emphysema, even in the absence of ILD changes at imaging, further suggesting that emphysema with mild fibrosis of alveolar walls at pathology on one hand, and overt CPFE syndrome on the other hand, may be part of a continuum of smoking-related lung disease.
Complications and Outcome
Making the diagnosis of the CPFE syndrome is highly relevant, because the outcome and risk of complications is distinct from that of either IPF or emphysema alone. The disease course of the CPFE syndrome is often characteristic, with emphysema preceding the onset of the pulmonary fibrosis in the majority of cases [7], and a dismal prognosis. PH may occur in about half of the cases after a median of 18 months following the diagnosis of CPFE [20] (Fig. 22.10). Survival is severely reduced, with a median of 12–18 months from the onset of confirmed precapillary PH. Lung cancer may occur after a median of 2 years after the diagnosis of CPFE (from 0.6 to 11.2 years in our series [10]).
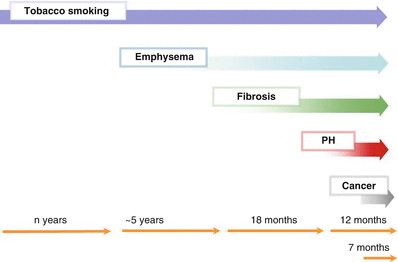
Fig. 22.10
Temporal Diagram of the main complications in tobacco-related CPFE syndrome. The median delay between emphysema and fibrosis is 5 years, and pulmonary hypertension may occur after a median of 18 months after the diagnosis of CPFE. Patients may die after a median of about 1 year from the diagnosis of pulmonary hypertension and a median of 7 months from the onset of lung cancer, with great inter-individual variability. PH pulmonary hypertension
Mortality
The overall median survival in CPFE is comprised between 2.1 and 8.5 years [7, 14–16, 18, 103, 129–131]. In CPFE and connective tissue disease, the overall survival was 100 % at 1 year, 94 % at 2 years, and 73 % at 5 years [21]. The main causes of deaths in patients with CPFE are represented by severe PH, intractable hypoxemia, pulmonary infection, and lung cancer. The relatively preserved lung volumes in CPFE may underestimate the severity of the pulmonary fibrosis.
Comparison of survival between patients with CPFE and IPF has been controversial [130], as results may vary according to: (1) diagnostic criteria for CPFE, with survival in CPFE possibly confounded by patients with pathology patterns other than UIP especially NSIP (i.e. non-UIP CPFE); for example, the proportion of patients with NSIP was higher in the CPFE group than in the control group in one study that found better survival in CPFE than in pulmonary fibrosis without emphysema [129]; (2) difficulties in controlling for the severity of fibrosis; (3) survival time bias, with presumably earlier diagnosis due to more severe symptoms in patients with CPFE as compared to IPF after adjustment for the severity of fibrosis; patients with CPFE more frequently have chronic bronchitis than those with IPF and may seek medical attention earlier [130], which may explain why fibrosis and emphysema are inversely correlated at the diagnosis of CPFE [105]; (4) the method used to handle transplantation [130]. A composite physiologic index predicts mortality in isolated IPF and may account for disease severity [4], however there are uncertainties regarding its use in CPFE syndrome [17]. In one series that defined CPFE as IPF (with a CT and/or pathological pattern of UIP) associated with emphysema, mortality was similar in patients with CPFE as compared to IPF after adjustment for the severity of fibrosis at imaging [18]. It should be emphasized that there is great variability of progression between patients with the CPFE syndrome, with individuals with slow or rapid progression of disease especially regarding the fibrosis (Fig. 22.11).
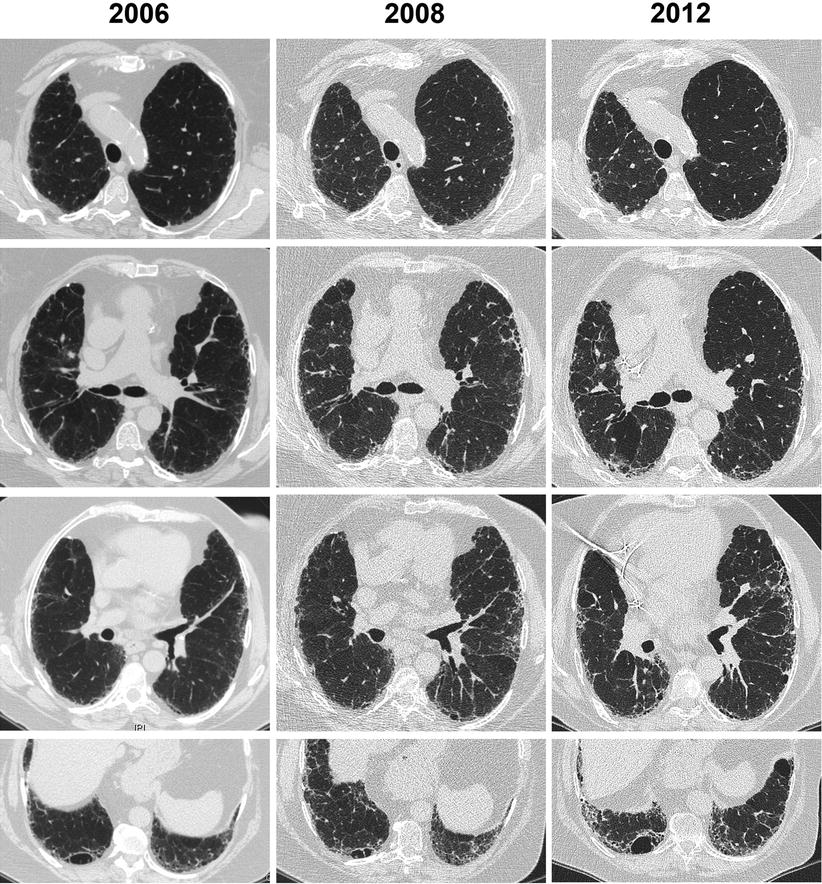
Fig. 22.11
Example of slow progression of disease in a woman with smoking-related CPFE syndrome (chest CT), who quit smoking at diagnosis. Only mild centrilobular emphysema is present in the upper lobes. Subpleural reticular changes predominate, which progress to a pattern of possible usual interstitial pneumonia. A thick-walled large cyst is present in the right lower lobe, which increases in size
Pulmonary Hypertension
The main determinant of prognosis in CPFE is represented by PH [7, 16], which develops in up to half of patients with CPFE [7]. PH was present at echocardiography in 47 % of patients in the seminal series of 61 patients [7]. In a cohort of 102 patients diagnosed with CPFE at our center after that publication, PH was present at right heart catheterisation in 48 % of patients with CPFE (Ahmad K and Cottin V, unpublished). When present, PH may cause dilation of the pulmonary arteries at chest radiograph and CT. Newer techniques including magnetic resonance imaging may contribute to the evaluation of PH in CPFE [132], however right heart catheterisation remains the gold standard to diagnose PH.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


