(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Hypertension is a major public health problem and is one of the most important risk factors for the development of atherosclerosis and vascular events. Although it is a multifactorial condition, hypertension is associated with oxidative stress and endothelial dysfunction leading to increased vascular resistance. These perturbations likely represent both a cause and a consequence of elevated levels of reactive oxygen (ROS) and nitrogen (RNS) species. Mitochondria are important sites of ROS production, and mitochondrial dysfunction, preceding endothelial dysfunction, might favor the development of hypertension. ROS production may also be induced by RNS, which inhibit the respiratory chain, and may be generated through the action of a mitochondrial NO synthase. Mitochondrial uncoupling proteins are involved in both experimental and human hypertension. Finally, an excessive production of ROS may damage mitochondrial DNA, with resultant impairment in the synthesis of some components of the respiratory chain and further ROS production, a vicious cycle that may be implicated in hypertension.
Introduction
Hypertension is a major public health problem and is a risk factor for atherosclerosis and vascular events [1]. Human arterial hypertension is a condition associated with oxidative stress and endothelial dysfunction [2–5]. Oxidative stress refers to processes characterized by an imbalance between the excessive formation of reactive oxygen (ROS) or nitrogen (RNS) species and limited antioxidant defense [6]. Like other risk factors of atherosclerosis and heart disease [7, 8], however, it remains uncertain whether elevated levels of ROS and RNS that initiate the development of hypertension as a consequence of the vasoconstriction induced by endothelial dysfunction are a consequence of hypertension itself or both (Fig. 15.1) [9]. A higher superoxide and hydrogen peroxide production has been observed in hypertensive patients [10]. Further, in untreated hypertensives, a decreased antioxidant superoxide dismutase (SOD) and glutathione peroxidase activity has been shown in comparison with controls [11]. On the other hand, in whole blood and in mononuclear cells from hypertensive subjects, although there were an increase in oxidative stress and a reduction in the activity of antioxidant mechanisms, they appear to be independent of the blood pressure values [3]. Overall, these data may indicate that factors other than blood pressure, such as a hyperadrenergic status, enhanced activity of angiotensin II, or hyperinsulinemia, may be responsible for the altered oxidative status in hypertensive patients.
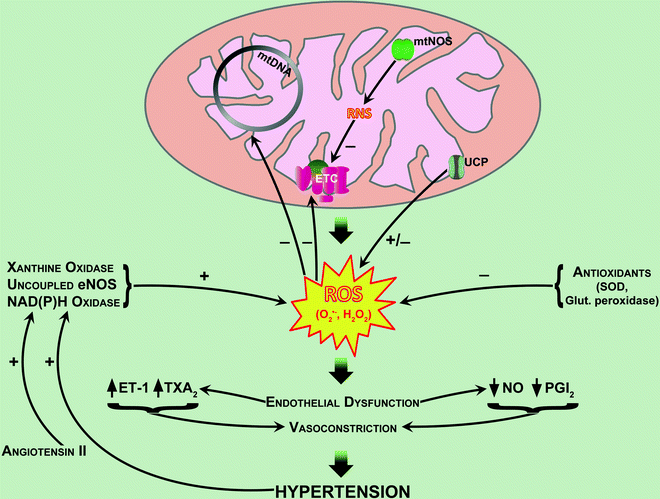
Fig. 15.1
The putative role of mitochondrial dysfunction in hypertension. See text for details. Abbreviations: eNOS endothelial nitric oxide synthase; ET-1 endothelin-1; ETC electron transport chain; glut peroxidase, glutathione peroxidase; H 2 O 2 hydrogen peroxide; mtDNA mitochondrial DNA; mtNOS mitochondrial nitric oxide synthase; NO nitric oxide; O 2•− superoxide anion; PGI 2 prostacyclin; RNS reactive nitrogen species; ROS reactive oxygen species; SOD superoxide dismutase; TXA 2 thromboxane A2; UCP uncoupling protein
Several enzymes are believed to be responsible for an increased production of ROS in the vasculature. These include NAD(P)H oxidase, NO synthase (NOS), lipoxygenases, cyclooxygenases, xanthine oxidase, and cytochrome P450 (CYP 450) enzymes. In particular, recent reports have clearly indicated that superoxide production induced by angiotensin II is regulated by NAD(P)H-dependent mechanisms [12, 13], suggesting that NAD(P)H oxidase might be the most important source of superoxide anion [14]. In a comparison of the superoxide-generating capabilities of vascular smooth muscle cells (VSMCs) derived from wild-type (p47phox(+/+), phagocyte oxidase) mice with those from mice that lack p47phox (p47phox(−/−), “knockout”), an essential component of the phagocyte NAD(P)H oxidase, p47phox was detected only in p47phox(+/+) VSMCs. p47phox-transduced p47phox(−/−), but not enhanced green fluorescent protein-transduced p47phox(−/−) VSMCs, generated significant levels of superoxide after stimulation by angiotensin II or platelet-derived growth factor-BB (PDGF-BB). Enhanced expression of recombinant p47phox in p47phox-transduced p47phox(−/−) cells correlated with superoxide production in these cells. These data provide direct functional evidence that an oxidase requiring the p47phox component mediates superoxide release from VSMCs in the blood vessel wall in response to angiotensin II or PDGF-BB [13].
Mitochondria represent both a major site of ROS production and a target of ROS action [15, 16]. Moreover, the ROS-induced instability of critical non-lipid molecules, such as mitochondrial DNA (mtDNA) that will be discussed in a later section [17], may alter oxidative phosphorylation, with a further increase in ROS production [18]. Molecular mechanisms of mitochondrial ROS production, the role of mitochondrial dysfunction, mitochondrial uncoupling proteins, and mitochondrial oxidative and DNA damage in hypertension will be reviewed in this chapter.
Mechanisms of ROS Generation and Mitochondrial Dysfunction
In aerobic metabolism, the energy of mitochondrial electron transport is converted into the high-energy phosphate bond of ATP via a multicomponent NADH dehydrogenase complex [19]. Molecular O2 is the final electron acceptor for cytochrome c oxidase or complex IV, the terminal component of the respiratory chain, and is ultimately reduced to H2O. However, a small quantity of O2 may not be completely reduced, as a leakage of single electrons can cause the reduction of oxygen to superoxide. The rate of ROS production depends on mitochondrial inner membrane potential, which can be depolarized by mitochondrial ATP-sensitive potassium channel openers [20]. Most superoxide and some hydroperoxy radicals are generated in complex I (NADH-coenzyme Q reductase) and complex III (ubiquinone-cytochrome c reductase) of the respiratory chain [21, 22]. Moreover, it has recently been shown that electrons derived from FADH2 (complex II substrate) can undergo reverse transport into complex I, possibly representing the primary source of superoxide production in mitochondria [23–25]. Accordingly, a variety of mitochondrial sites of superoxide production have been identified, including several respiratory complexes as well as individual enzymes [26].
The simultaneous formation of superoxide and nitric oxide (NO) radicals produces peroxynitrite, a strong oxidant and nitrating agent. It had been proposed that a putative new isoform of mitochondrial NO synthase (mtNOS) could be located in the mitochondrial matrix [26–29]. As a matter of fact, the existence of mtNOS is by no means uncertain [28], since NOS may not be contained within the mitochondrial matrix but rather attached to the outer membrane of mitochondria [28, 30]. However, even if mtNOS is not localized in the matrix, it may have a specific connection with mitochondria, which might play a physiological role in the regulation of enzyme activity. Nevertheless, changes in brain mtNOS expression and activity could not be found in spontaneously hypertensive rats (SHRs) [27]. Additional studies will be required to further evaluate the physiological significance of vascular mtNOS in hypertension [31].
In mitochondria, peroxynitrite, NO, and other RNS can interact with complexes I, III, and IV of the respiratory chain [32]. In particular, the key site for the interaction of NO with mitochondria is at cytochrome c oxidase, which is competitively inhibited, with subsequent reversible inhibition of O2 consumption and ATP synthesis [29]. The interaction between RNS and mitochondria can also exert significant effects on both mitochondrial and cellular signaling events implicated in apoptosis and redox regulation of gene expressions [32]. Furthermore, mtDNA damage by ROS, generated as a consequence of the RNS-induced impairment in oxidative phosphorylation, could play a significant role in the pathogenesis of hypertension [33, 34], as will be reviewed in a subsequent section.
Angiotensin II is a powerful inductor of oxidative stress, and the production of superoxide in response to angiotensin II in vascular tissues is mainly mediated by NAD(P)H oxidase [14, 35]. Taylor and coworkers have shown that NAD(P)H oxidase also induces oxidative stress in the renal medulla of Dahl salt-sensitive rats, thereby contributing to the development of salt-induced hypertension [36]. In addition, Kimura et al. have shown that angiotensin II is able to stimulate mitochondrial ROS generation through the activation of mitochondrial ATP-sensitive potassium channel openers [37]. The mitochondrial dysfunction in the kidney observed in the SHRs is improved by angiotensin II blockade, further suggesting a role for angiotensin II in the induction of oxidative stress [38]. On the other hand, ROS generated by mitochondria cannot influence vascular contractility during the chronic phase of angiotensin II-induced hypertension [37].
Mitochondrial dysfunction has now been implicated in both human and experimental hypertension [10, 39]. It has been shown that hypertension is associated with a deterioration of mitochondrial energy production both in SHRs [32, 40] and in mice [41]. Furthermore, in experimental hypertension, mitochondrial energy deficiency [33] and a decreased activity of complex IV have been observed in the hypertrophied myocardium from SHRs [42]. In these animals, there is also a decrease in the activity of inorganic phosphate translocator [43], together with a decrease in ATP production [40]. These data therefore confirm the results first obtained in 1990 by Das and Harris [44], who reported that cardiomyocytes from normotensive rats responded to increased energy demand caused by electrical stimulation or treatment with positive inotropic agents, by increasing their ATP synthase capacity up to twofold, whereas cells from SHRs were unable to control their ATP synthase in this manner. In experimental neurogenic hypertension, feedforward depression of mitochondrial electron transport chain appears to function by superoxide anion and hydrogen peroxide in rostral ventrolateral medulla, a brain stem site that maintains sympathetic vasomotor tone and contributes to oxidative stress and neural mechanism of hypertension [39]. As discussed earlier, deterioration of mitochondrial energy production may play a role in the pathogenesis of hypertension in both SHRs [32, 40] and mice [41]. Mitochondrial energy deficiency [33] and a decreased activity of complex IV have also been observed in the hypertrophied myocardium from SHRs [42]. In these animals, there is also an abnormal transport of inorganic phosphate in left ventricular mitochondria [43]. Overall, these data indicate that some defects in the regulation of mitochondrial ATP synthase activity occur in the cardiomyocytes of SHRs.
Mitochondrial energy deficiency in conjunction with calcium overload may play a role in the pathogenesis of arterial hypertension [33]. As discussed earlier, a decrease in mitochondrial energy metabolism and abnormality of calcium metabolism has been reported in hypertrophied myocardium of spontaneously hypertensive rats [40, 42]. Mitochondrial antioxidant system is also important in protection against hypertension because arterial blood pressure increased with aging or high-salt diet in SOD2-deficient mice [45]. Indeed, wild-type (SOD2(+/+)) and partial SOD2-deficient (SOD2(+/−)) mice had similar blood pressures at 6–7 months of age, but at 2 years SOD2(+/−) mice had higher blood pressure. Oxidative stress, renal interstitial T-cell and macrophage infiltration, tubular damage, and glomerular sclerosis were all significantly increased in 2-year-old SOD2(+/−) mice. High-salt diet induced hypertension in 6-month-old SOD2-deficient mice but not in wild-type mice. In conclusion, partial SOD2 deficiency results in oxidative stress and renal interstitial inflammation, changes compatible with accelerated renal senescence and salt-sensitive hypertension [45]. Mild respiratory uncoupling in arterial smooth muscle cells increased oxidative stress, hypertension, and dietary atherosclerosis in mice [41]. Thus, as will be discussed in further details in the next section, in mice with doxycycline-inducible expression of uncoupling protein-1 (UCP1) in the artery wall, expression of UCP1 in aortic smooth muscle cells caused hypertension and increased dietary atherosclerosis without affecting cholesterol levels. UCP1 expression also increases superoxide production and decreases the availability of NO, evidence of oxidative stress [41]. These results again provide proof of principle that inefficient metabolism in blood vessels can cause vascular disease. Recently, it was shown that a mutation in mitochondrial tRNA results in hypertension, hypercholesterolemia, and hypomagnesemia [46]. Each phenotype was transmitted on the maternal lineage with a pattern indicating mitochondrial inheritance. Analysis of the mitochondrial genome of the maternal lineage identified a homoplasmic mutation substituting cytidine for uridine immediately 5′ to the mitochondrial transfer RNAIle anticodon. Because cholesterol concentration and blood pressure increase at approximately 30 years of age in these patients, it has therefore been hypothesized that this gene mutation interacts with environmental or age-related decline in mitochondrial function in the development of hypertension and hypercholesterolemia [47]. Taken together, these findings indicate a potential role of mitochondrial dysfunction in hypertension. The putative role of mitochondrial dysfunction in hypertension has been recently reviewed [48] and is illustrated graphically in Fig. 15.1.
The Role of Mitochondrial Uncoupling Proteins
A significant association of hypertension with mitochondrial uncoupling proteins (UCPs) has been reported in both experimental and human hypertensive states. To test the hypothesis that inefficient metabolism in blood vessels promotes vascular disease, mice with doxycycline-inducible expression of UCP1 in the artery wall were generated. UCP1 expression in aortic smooth muscle cells caused hypertension and increased dietary atherosclerosis without affecting cholesterol levels. UCP1 expression also increases superoxide production and decreases the availability of NO, evidence of oxidative stress [41]. These results provide proof of principle that inefficient metabolism in blood vessels can cause vascular disease. Douette et al. [49] investigated how active and regulated recombinant UCP1 expressed in yeast at approximately 1 and approximately 10 μg/mg of total mitochondrial proteins induced changes in the mitochondrial proteome and in oxygen free radical production. Using two-dimensional differential in-gel electrophoresis (2D-DIGE), they found that most of the proteins involved in the response to ectopically expressed UCP1 are related to energy metabolism. These investigators also quantified the cellular H2O2 release in the absence or in the presence of UCP1. UCP1 had a dual influence on free radical generation. On one hand, free fatty acids (FFA) activation of UCP1 induced a decrease in superoxide anion production, demonstrating that a decrease in ROS generation is an obligatory outcome of UCP1 activity. On the other hand, an increase in UCP1 content was associated with an increase in the basal release of superoxide by mitochondria as a consequence of the overall increase in oxidative metabolism. UCP2 content in kidney mitochondria of SHRs is lower than in controls without hypertension [38]. Indeed, a major function of this protein may be to increase the proton conductance across the inner mitochondrial membrane, thereby reducing superoxide production [50].
A common polymorphism of the UCP2 gene appeared to be associated with hypertension in a Japanese population and with hypertension and obesity in Caucasians [51]. The frequency of the minor A allele was significantly higher in Japanese than in Caucasians (48.9% vs. 37.2%, P = 0.01). In contrast to the significant association with obesity in Caucasians, the polymorphism was not associated with obesity in the Japanese. The polymorphism, however, was significantly associated with hypertension in Japanese (frequency of A allele: 51.8% in hypertensives vs. 46.6% in normotensives, P < 0.05). No significant differences, however, were observed in body mass index (BMI), fasting insulin level, or insulin sensitivity between patients with the different genotypes.
The Mitochondrial DNA and Oxidative Damage
Excessive production of ROS and RNS in mitochondria can damage mitochondrial DNA (mtDNA), with decreased energy production, additional generation of ROS, and enhancement of the cellular signals capable of initiating hypertension as well as atherosclerosis [52]. An excessive production of ROS in mitochondria may damage mtDNA, which is located close to the inner membrane and is not protected by histone proteins, as is the case for nuclear DNA (nDNA) [53]. Under normal conditions in mitochondria, there is a balance between ROS formation and degradation. In pathological circumstances, the antioxidant defenses and mtDNA-repairing enzymes become insufficient, resulting in oxidative stress and leading to mtDNA damage. This may have relevant consequences because although 95% of mitochondrial proteins are encoded by nuclear DNA, mtDNA contains genes coding 13 components of the respiratory chain, subunits of NAD(P)H oxidase, cytochrome c oxidase and cytochrome b [26, 34].
NO can damage mtDNA to a greater extent than nuclear DNA [52]. To determine whether enhanced repair resulting from augmented expression of human DNA repair enzyme, 8-oxoguanine DNA N-glycosylase 1 (hOGG1), could also protect against the deleterious effects of NO, HeLa TetOff/MTS-OGG1-transfected cells were utilized to conditionally express hOGG1 in mitochondria. The effects of additional hOGG1 expression on repair of NO-induced mtDNA damage and cell survival were evaluated. These cells, along with vector transfectants, in either the presence or absence of doxycycline (Dox), were exposed to NO produced by the rapid decomposition of 1-propanamine, 3-(2-hydroxy-2-nitroso-1-propylhydrazino) (PAPA NONOate). Subsequent functional studies revealed that cells expressing recombinant hOGG1 were more proficient at repairing NO-induced mtDNA damage, which led to increased cellular survival following NO exposure. Moreover, the results demonstrate that conditional expression of hOGG1 in mitochondria decreases NO-induced inhibition of ATP production and protects cells from NO-induced apoptosis [52]. The overall accumulation of mtDNA mutations can cause cellular dysfunction by altering ATP generation, and Ca2+ homeostasis, inducing further oxidative stress, a defective turnover of mitochondrial proteins, and the production of defective protein subunits [54]. The processes in mitochondria, although co-regulated with the energy demand of the host cell and partially with the cell cycle, occur at much faster rates than those in the hosts. Consequently, each mitochondrion contains two to ten copies of its genome, and each cell encompasses hundreds to thousands of copies of mitochondria. The faster rate of mtDNA replication, however, is not without consequences, particularly, in an environment that is high in ROS. The rare error rates of DNA replication and editing enzymes increase in the presence of oxidation-modified nucleotides and enzymes. Hence, mtDNA has 16 times higher mutation accumulation rate than the nDNA [55]. A vicious cycle may therefore be generated, which leads to the progressive accumulation of mutations and oxidative damage to mtDNA [56].
Mitochondrial DNA mutations have been demonstrated in African Americans with hypertension-associated end-stage renal disease [57]. Moreover, as discussed earlier, it has been shown that a mutation in mtRNA is associated with hypertension, hypercholesterolemia, and hypomagnesemia [46]. Mitochondrial DNA can also be affected by inherited defects of the genes-encoding components of the oxidative phosphorylation, with possible additional contributions to the pathophysiology of the hypertrophied myocardium in stroke-prone SHRs (SHRSPs) [58]. Morphologically, the mitochondrial size showed a wider range of distribution in stroke-prone SHRs at both prehypertensive and hypertensive stages compared to those in age-matched normotensive Wistar-Kyoto (WKY) rats. Isocitrate dehydrogenase activity, but not SOD activity, was higher in the young SHRSPs, whereas both enzyme activities were lower in the mature stroke-prone SHRs than in the age-matched WKYs. Because the mtDNA codes for many specific proteins of the electron transport chain, it is plausible that mutations or deletions in mtDNA could result in decreased energy production and additional generation of ROS, thus enhancing the cellular signals capable to initiate hypertension as well as atherosclerosis, apoptosis, and necrosis [52].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


