(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Mitochondria are dynamic organelles displaying considerable structural complexity and diversity in terms of the folding of their inner membrane in particular. The elaborate topology of the inner membrane, which provides sites for assembly of OXPHOS complexes, is highly dynamic and regulated to optimize mitochondrial performance in response to metabolic demands. Cardiac mitochondria display a unique arrangement within cardiomyocytes and are classified, depending on their intracellular location, as intermyofibrillar, subsarcolemmal, or perinuclear. Structural differences, mainly in the morphology of the cristae, underlay functional differences between these mitochondrial subpopulations. Mitochondria communicate with the cell via numerous channels. K+, Na+, Ca2+, H+ as well as Pi, nucleotides, and various proteins cross the mitochondrial membranes in a highly regulated manner to orchestrate a variety of processes ranging from ATP synthesis and mitochondria dynamic organization to cell survival and death. Unfortunately, the molecular identity of the majority of mitochondrial channels remains unknown. Cardiac mitochondria undergo movement along microtubules and constant remodeling from elongated interconnected networks to fragmented disconnected mitochondria. These processes are governed by complex, evolutionary conserved, machineries of mitochondrial transport and fusion and fission. A tight balance of fusion and fission is critical for the maintenance of mitochondrial function. The molecular mechanism and regulation of mitochondrial dynamics are best understood in yeast and only begin to be appreciated in humans. However, emerging evidence suggests that perturbations in mitochondrial dynamics can be detrimental to the myocardium and may play essential roles in the pathogenesis of cardiac disorders, such as ischemia–reperfusion injury, cardiomyopathies, and heart failure. Future progress in our understanding of crosstalk between mitochondrial morphology and physiological function is likely to provide novel therapeutic targets for the more efficient treatment of cardiovascular disorders.
Introduction
The human heart consumes approximately 6 kg of ATP every day to maintain constant rhythmic contractions pumping almost 10 tons of blood to ensure oxygenation of all tissues of the body. ATP utilized by the myocardium is generated mainly by oxidative phosphorylation (OXPHOS) in mitochondria. Cardiac mitochondria occupy 20–30% of the cardiomyocyte volume highlighting their critical role in these high-energy-consuming cells. Permanent contractions require specific structural organization of mitochondria within cardiomyocytes providing a bioenergetic basis for cardiomyocyte function. Moreover, structure, number, and metabolism of cardiac mitochondria can be changed significantly in response to various stimuli.
Mitochondria were first described by the Swiss anatomist Rudolf Albrecht von Kolliker in 1857. However, only in 1898, the German microbiologist Carl Benda introduced the term “mitochondria” (from the Greek words “mitos,” thread, and “chondros,” grain) reflecting the typical shapes of mitochondria identified by light microscopy [1, 2]. In the first half of the last century, the Belgian biochemist Albert Claude, who demonstrated that isolated mitochondria were able to catalyze respiration, concluded “that mitochondria may be considered as the real power plants of the cell” [3]. Since that revolutionary discovery, tremendous progress has been made in deciphering the molecular structure and mechanisms of the mitochondrial machineries involved in energy metabolism. Moreover, it is now widely recognized that mitochondria are not only the cellular “powerhouse” generating ATP, but they are also key regulators of oxidative stress response and reactive oxidative species (ROS) generation, cell death, various signaling pathways, and intracellular Ca2+ homeostasis.
In this chapter we will discuss the mitochondrial arrangement in cardiomyocytes and the morphology and molecular structure of these complex organelles. Finally, recently discovered highly regulated dynamic changes, processes of fusion, fission, and trafficking of mitochondria will also be reviewed.
Mitochondrial Arrangement in Cardiomyocytes
In mammalian cardiomyocytes, three subpopulations of mitochondria have been described based on differences in their location, morphology, or methods of isolation [4–6]. Intermyofibrillar mitochondria (IMFM) are localized deeper in the cell, longitudinally packed between the sarcomere myofibrils, while subsarcolemmal mitochondria (SSM) are clustered in contact with the sarcolemma. Perinuclear mitochondria (PNM) are typically localized nearby the nucleus and are significantly smaller and have more rounded shape compared to the larger and complex-shaped IMFM. Furthermore, isolation of tightly packed IMFM is very difficult and required additional nagarse treatment, whereas SSM can be easily isolated by polytron treatment of the cardiac tissue [7, 8].
In addition, IMFM and SSM often differ in the structure of their cristae: IMFM have tubular cristae, whereas SSM have lamelliform cristae [4, 9] (Fig. 3.1). However, cardiac mitochondria are dynamic organelles, and described differences in morphology of three mitochondrial subpopulations are highly variable depending on physiological and pathological conditions [9].
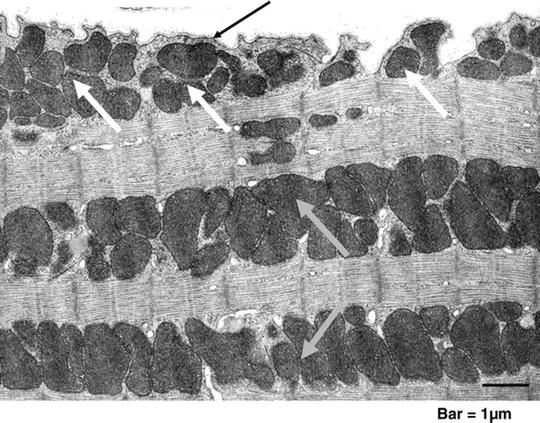
Fig. 3.1
Cardiac mitochondria. Transmission electron micrograph of mouse myocardium. Clusters of subsarcolemmal mitochondria (SSM, white arrows) are situated immediately beneath the sarcolemma (black arrow); longitudinal rows of mitochondria that are located within the contractile apparatus (myofibrils) are interfibrillar mitochondria (IFM, grey arrows) ([10] with permission from Oxford University Press)
Importantly, purified IMFM have different biochemical characteristics compared to other cardiac mitochondria subtypes: they have almost 50% higher rates of OXPHOS, significantly higher cytochrome c levels and up to threefold higher capacity for Ca2+ uptake than SSM [7, 11, 12]. The latter feature explains why Ca2+ overload leads to cytochrome c release, a prelude to apoptosis, mainly from SSM [13].
Functional differences between isolated cardiac mitochondrial subpopulations have been confirmed by observations on intact mitochondria. Hypoxia induced the swelling of all cardiac mitochondria; however, only IMFM and PNM demonstrated a marked increase in area and number of cristae, and also the number of PNM was significantly elevated [14]. Intriguingly, ischemia/reperfusion (I/R) in rats results in mitochondrial damage (decreased OXPHOS) of both SSM and IMFM, while in rabbits it affects only SSM; irrespective of species the net OXPHOS was significantly reduced [15, 16].
Internal Structure of Mitochondria
Great advances in imaging techniques, the development of high-resolution fluorescent microscopy, electron microscopy (EM), and EM tomography, during the last 50 years have revolutionized the field of mitochondria research. Transmission EM has revealed mitochondria as double-membrane-enclosed organelles with an inner membrane forming numerous invaginations, cristae. The following mitochondrial subcompartments are typically recognized: the mitochondrial outer and inner membranes (MOM and MIM, respectively) with cristae form two aqueous compartments—the space between the MOM and MIM called the intermembrane space (IMS), and the space enclosed by the MIM called the mitochondrial matrix. In addition, the regions of close contacts of the MOM and MIM are called contact sites, while the regions where cristae connect to the MIM are called cristae junctions (Fig. 3.2) [17–19].
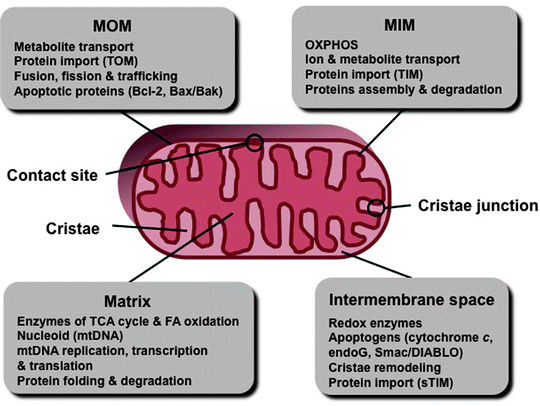
Fig. 3.2
Schematic representation of the mitochondrial compartments. Major compartment-specific processes and factors are shown. endoG endonuclease-G, FA fatty acids, MIM mitochondrial inner membrane, MOM mitochondrial outer membrane, mtDNA mitochondrial DNA, sTIM soluble TIM proteins, TCA tricarboxylic acid, TIM translocase of the MIM, and TOM translocase of the MOM
Development of conventional methods of specimen preparation for EM and especially the newly emerging cryo-EM tomography, which uses quickly frozen samples and devoids artifacts induced by chemical fixation, dehydration, and staining, provides excellent preservation of mitochondrial ultrastructure [20–26]. These novel techniques have confirmed mitochondrial compartmentation and provided further insights into its ultrastructural organization.
Although mitochondrial morphology (e.g., cristae structure and arrangement) depends on physiological conditions, a careful analysis demonstrates that mitochondria from various sources reconstructed in situ possess common typical structural characteristics (Fig. 3.3) [17, 27, 28]. Under normal physiological conditions, fungal, plant, and animal mitochondria are formed by a smooth MOM, which envelops the MIM with numerous invaginations, the cristae, enclosing a protein-rich matrix. The average distance between the MOM and MIM is 20 nm, narrowing to 14 nm at contact sites with no resolved space between the membranes. All tubular or lamellar cristae connect to the MIM on one side through narrow tubular cristae junctions with a diameter of approximately 28 nm and of variable length (Fig. 3.3) [17, 29–33].
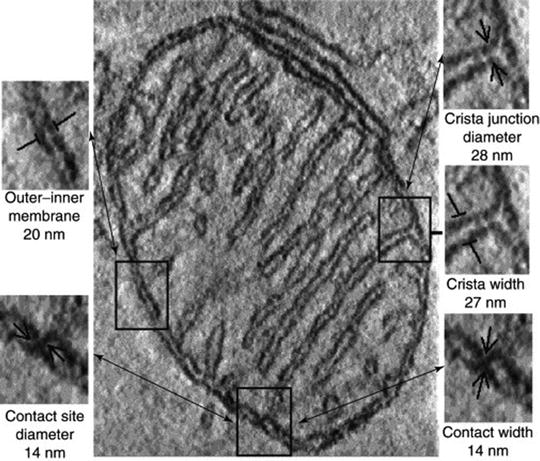
Fig. 3.3
A single section through the 3D tomogram of the mitochondrion. Examples of the various structural features are outlined, identified, and enlarged to the left and right, along with their dimensions averaged from the complete tomograms of multiple mitochondria reconstructed in situ in neural tissue, brown adipose tissue, and Neurospora crassa ([17] with permission from Elsevier)
Pioneering electron microscopic studies on mitochondrial ultrastructure by Palade and Sjostrand led to two models. According to the “septa model” proposed by Sjorstrand, the mitochondrial matrix is divided by the MIM into many distinct compartments forming septa spanning through the matrix [34]. Palade’s model, known as the “baffle model,” recognizes the internal membrane compartments, cristae mitochondriales, but they are formed by baffles protruding into the matrix with paths around them rather than septa [35, 36]. According to the widely accepted current model, the cristae are not random involutions of the MIM, but they are complex tubular and lamellar structures, which connect to the MIM via narrow tubular regions, cristae junctions [17, 27, 29]. Cristae significantly increase the surface area of the MIM providing sites for assembly of OXPHOS complexes. Consistently, their numbers are markedly elevated in highly respiratory-active cardiomyocytes (i.e., threefold more than found in hepatocytes). Importantly, this structural design is highly flexible and dynamic: the shape of the MIM can rapidly change and adjust in response to metabolic demands, underlying the plasticity of mitochondria [37, 38].
Mitochondrial Outer and Inner Membranes
The functional flexibility of the mitochondrial structure is determined by a fluid phospholipid bilayer of the mitochondrial membranes. Mitochondrial membranes contain the major classes of phospholipids characteristic to all cellular membranes, such as phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, phosphatidylserine, phosphatidic acid, and phosphatidylglycerol. In addition to these common membrane phospholipids, mitochondrial membranes contain cardiolipin (CL), which is found predominantly if not exclusively in mitochondria and largely in the MIM [39].
Like most biological membranes, the MOM has about 1:1 phospholipid to protein ratio. Approximately 4% of total mitochondrial proteins are located in the MOM [40]. MOM proteins represent two types of proteins: integral membrane proteins, containing transmembrane domains, and peripheral membrane-associated proteins. They contribute to various essential functions, such as import of fatty acids and proteins into mitochondria, exchange of hydrophilic solutes between the IMS and cytoplasm, and multiple interactions between mitochondria and other organelles by creating docking sites for the interacting proteins [41–44]. The most abundant protein in the MOM, the voltage-dependent anion channel (VDAC), also known as porin, facilitates the transport of ATP/ADP across the outer membrane [45, 46].
In contrast to the majority of biological membranes including the MOM, the MIM is characterized by a higher protein content exhibiting a protein to lipid ratio, more than 3:1 by weight. Proteins localized in the MIM account for about 21% of the total mitochondrial protein [40]. Since the MIM is highly impermeable to solutes and metabolites, their transport in and out the mitochondrial matrix is mediated by specific membrane-spanning carrier proteins. The translocase of the inner membrane (TIM) in concert with translocase of the outer membrane (TOM) mediates protein import into the mitochondrial matrix.
The MIM also contains three major mitochondrial K+ channels: the ATP-sensitive K+ channel (mitoKATP), the voltage-dependent K+ channel (mitoKV), and the Ca2+-activated K+ channel (mitoBKCa). Finally, numerous invaginations of the MIM, the cristae, of highly changeable number and shape contain the electron transport chain complexes I–IV and the F0F1 ATP synthetase (complex V) (see Chap. 5).
Since the first demonstration of physical contacts between the MOM and MIM, so-called contact sites, by Hackenbrock, they have been implicated in various mitochondrial processes, such as metabolite and protein transport, bioenergetics, and apoptosis [47–51]. Initial observations of numerous contact sites were based on transmission EM of chemically fixed and dehydrated mitochondrial specimens; however, more recent quick-freeze, deep-etch EM and electron tomographic reconstruction of unfixed, frozen-hydrated mitochondria and tissue sections, although have confirmed the presence of contact sites, have demonstrated fewer numbers [52–55]. The presence of the 10–15 nm bridging IMS particles at contact sites suggests that they may represent multiprotein complexes serving various functions. However, it is currently unclear whether they link physically the MOM and MIM. Moreover, contact sites are enriched for CL, which facilitates action of protein complexes involved in transport of small molecules and proteins [56].
Cardiolipin
CL was initially isolated from bovine heart and was subsequently demonstrated in ATP-producing membranes from protists, prokaryotes, and eukaryotes [57, 58]. CL represents a relatively small fraction (approximately 16% in rats) of the cardiac phospholipids; however, it is critical for metabolite and protein transport, for OXPHOS, and for the apoptotic signaling cascades [59–63]. In the normal mammalian heart, CL has been found only in mitochondria, predominantly in the MIM, colocalizing with protein assemblies involved in OXPHOS and protein and metabolite transport.
CL is a glycerophospholipid composed of two diacylglycerol phosphate residues and four fatty acid chains. Although CL was the first membrane phospholipid which composition was determined, it was the last of major phospholipids to be characterized stereochemically [63]. The titration curve of CL displays two pK’s at ∼4.0 and 8.0. The high pK 2 implies that in a bilayer at neutral pH the headgroup has a single negative charge and the headgroup conformation is stable when one of the headgroup phosphates picks up proton from solution. Thus, the bicyclic ring system created by hydrogen bonding of phosphates to the hydroxyl functions as a proton trap in the headgroup domain (Fig. 3.4) [64]. This unique molecular structure of CL facilitates proton-pumping generation of ΔΨm, which is required for ATP synthesis.
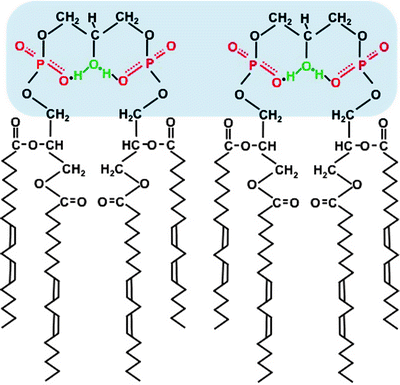
Fig. 3.4
Scheme of cardiolipin structure in a lipid bilayer. The symmetrical linoleate side chains stabilize the headgroup’s resonance structure, and a proton is trapped. The headgroups are marked by blue shadow
In mammalian heart, four fatty acyl side chains of CL are present, predominantly (up to 80% in humans) by linoleic acid (C18:2). Moreover, the linoleic acid chains in functional CL are arranged in a symmetrical manner (Fig. 3.4) [65]. This chain symmetry contributes to stabilization of the bicyclic headgroup structure. It also facilitates association between the headgroups of adjacent CL molecules forming highly dynamic CL domains within the MIM [63, 66]. Such molecular organization highly increases efficiency of proton transfer from ETC complexes to the F0F1 ATP synthase during ATP synthesis.
The importance of the symmetrical structure of CL was first highlighted by the discovery of the Barth syndrome characterized by cardiomyopathy, neutropenia, and increased blood levels of 3-methylglutaconic acid [67, 68]. This genetic X-linked recessive disease is caused by defective CL-remodeling enzyme, tafazzin, resulting in loss of the symmetric tetralinoleoyl CL and leading eventually to mitochondrial dysfunction [69–71]. Furthermore, alterations in the linoleic acid content and symmetry of CL have been associated with acute myocardial I/R injury, heart failure, ageing, and diabetes [61].
CL is synthesized in mitochondria. Its precursor, phosphatidic acid, is formed in the MOM and then migrate to the MIM, where in four enzyme-catalyzed steps the nascent tetra-acyl CL, not enriched for linoleate, is synthesized [61, 72]. This newly synthesized CL is remodeled to the linoleate-enriched cardiac CL by not fully understood mechanism. Two mitochondrial enzymes, monolysocardiolipin acyltransferase and tafazzin, and one ER enzyme, acyl-Coenzyme A, have been proposed to mediate CL remodeling. [58, 61, 71, 73] Defects in CL remodeling result in impaired mitochondrial function leading eventually to apoptosis, heart failure, or cardiomyopathy in experimental and clinical settings [70, 74, 75].
One of the most prominent features of CL is its capacity to bind non-covalently to the majority, if not all, proteins of the MIM. Among the proteins, which bind CL with high affinity, are the respiratory complexes I, III, IV, and V, the mitochondrial carriers (ADP/ATP carrier [ANT] and phosphate carrier [PiC]) and the IMS-located mitochondrial creatine kinase (mtCK), nucleotide diphosphate kinase (NDPK-D) and cytochrome c [76–79]. Moreover, although unlike ANT and PiC, uncoupling protein UCP1, which also belongs to the mitochondrial carrier family, has lower affinity to CL; CL attenuates the inhibitory effect of nucleotide binding on the UCP1 uncoupling activity and thereby contributes to its regulation [77].
CL within the MIM not only facilitates formation of OXPHOS complexes, but is also essential to their assembly into higher order supercomplexes [80, 81]. Similarly, CL is involved in association of the inner membrane ANT, with mtCK and NDPK-D facilitating metabolite transport (channeling) and regulating thereby mitochondrial bioenergetics [79]. CL also participates in tafazzin-complex V assemblies [82]. Moreover, numerous interactions of CL with proteins contribute to formation of membrane domains, CL clusters, that stabilize the curved regions of mitochondrial membranes [83].
In addition to the well established role for CL in the mitochondrial ATP synthesis, emerging evidence suggests that it contributes to mitochondria-mediated apoptosis. CL has been demonstrated to interact specifically with multiple apoptotic proteins, such as Bcl-2 family members, Bid, cytochrome c, and caspase-8 [62, 84]. During apoptosis, CL content reduces due to its degradation to monolyso-CL (MLCL), which appears to act as a signal of mitochondrial impairment [85, 86]. Increased MLCL translocates to the MOM through contact sites and contributes there to the recruitment of pro-apoptotic Bax/Bak and tBid leading to cristae remodeling and permeabilization of the MOM [87, 88]. Degradation and oxidation of CL in the MIM result in a decrease in cytochrome c binding to the MIM favoring further cytochrome c release into the cytosol [89–91].
It has been shown recently that the presence of CL in the MOM is also required for another critical apoptosis event—the recruitment of caspase-8 to mitochondria, which is necessary for its processing and activation during death receptor-induced apoptosis [92]. The mechanism of the CL-caspase-8 association and a possible role of other mitochondrial proteins in this process remain to be determined.
Thus, CL is emerging as a critical integrator, which orchestrates numerous mitochondrial processes ranging from the structural organization of mitochondrial membrane and ATP synthesis to metabolite and protein transport and apoptosis.
Mitochondrial Permeability Transition Pore
The mitochondrial permeability transition pore (MPTP) is a nonspecific channel permeable for most small molecules up to 1.5 kDa, which mediates the increases in the permeability of mitochondria leading to cardiomyocyte death and contributing to cardiovascular disease (CVD) [93–96]. MPTP was originally described in isolated mitochondria by Hunter and colleagues more than 30 years ago [97–100]. Since the pore is permeable to molecules of <1.5 kDa, its opening induced by Ca2+ overload accompanied by adenine nucleotide depletion, elevated phosphate concentrations, and oxidative stress (OS), conditions associated with I/R, results in failure to maintain a barrier to protons. The resulting dissipation of ΔΨm leads to inhibition of ATP synthesis and eventually to bioenergetic failure of the cardiomyocytes [93, 101, 102].
MPTP opening also causes organelle swelling and subsequent rupture of the MOM leading to the release of mitochondrial apoptogens, most notably cytochrome c, Smac/DIABLO, apoptosis-inducing factor (AIF), and endonuclease-G [103]. Ultimately these perturbations result in apoptotic and/or necrotic death of the cardiomyocytes [93, 104–106]. Of note, the main pathway causing the release of apoptogenic factors from the mitochondrial IMS is triggered by Bak/Bax and Bid promoting specific permeabilization of the MOM (see Chap. 11).
Despite appreciation of the critical role of MPTP, the precise molecular structure of the pore remains a mystery. Original models based on intensive biochemical and pharmacological investigations have proposed that MPTP spans both mitochondrial membranes and consists of VDAC in the MOM, the adenine nucleotide translocase (ANT) in the MIM, and cyclophilin D (CypD) in the matrix (Fig. 3.5a) [107–110]. Contact sites appear to be the predominant locations of VDAC–ANT–CypD complexes.
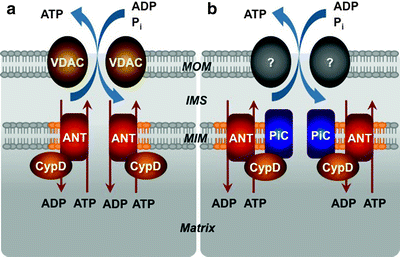
Fig. 3.5
Models for the molecular structure of the mitochondrial permeability transition pore (MPTP). (a) The initial model has proposed that the MPTP spans both mitochondrial membranes and consists of voltage-dependent anion channel (VDAC) in the mitochondrial outer membrane (MOM), adenine nucleotide translocator (ANT) in the mitochondrial inner membrane (MIM) and cyclophilin D (CypD) in the matrix. (b) According to the current model, VDAC is no longer a component of the MPTP; the MOM component is not known and may not even be necessary. The MIM located mitochondrial phosphate carrier (PiC) is added to the model as a candidate for the pore-forming subunit, while ANT and CypD represent regulatory subunits of the MPTP. IMS intermembrane space
Voltage-Dependent Anion Channel
VDAC, also referred to as porin, is the most abundant protein in the MOM involved in ATP/ADP transport across the membrane [45, 109, 111, 112]. Three isoforms, VDAC1, VDAC2, and VDAC3, are structurally and functionally homologous members of the VDAC family [45, 46]. Several biochemical studies have suggested VDAC as an essential component of the MPTP. Anti-VDAC antibodies and VDAC1-binding compound, Ro 68-3400, have been demonstrated to block Ca2+-induced mitochondrial permeabilization and cytochrome c release in isolated mitochondria [113, 114]. VDAC along with ANT have been pulled down by a GST-CypD fusion protein from mitochondrial lysates, and reconstitution of the VDAC–ANT–CypD complex has restored a Ca2+-dependent channel resembling the MPTP [115].
However, new evidence has seriously challenged the conclusions of these studies. Halestrap’s laboratory has demonstrated that a MPTP-like channel can be reconstituted in the absence of VDAC [116]. Moreover, it has been reported that the putative VDAC inhibitory agents are not totally specific: anti-VDAC antibodies recognize other non-VDAC proteins, and radioactive Ro 68-3400 labels another protein in VDAC1−/− mitochondria [46, 117]. Finally, it has recently been demonstrated that cells essentially deficient for all three VDAC are sensitive to Ca2+-induced mitochondrial permeabilization and cell death [118]. Interestingly, cells deficient in all VDAC isoforms have been more sensitive to OS-induced cell death suggesting a cytoprotective role for VDAC [118]. Thus, although VDAC is not an essential subunit of the MPTP, it appears to play a role in the maintenance of metabolite flux across the MOM during stress and in the mitochondrial targeting of protective proteins such as Akt, hexokinase, and protein kinase Cε [119–123].
Adenine Nucleotide Translocase
Another potential component of the MPTP is ANT, which belongs to the SLC25 family of the transmembrane mitochondrial carriers promoting ATP/ADP exchange across the MIM [124]. In mammals, three homologous ANT isoforms have been described. ANT1 is predominantly expressed in cardiac and skeletal muscle, while ANT2 and ANT3 are ubiquitously expressed; ANT3 has been found in humans but lacking in rodent; they are encoded by the SLC25a4, SLC25a5, and SLC25a6 genes, respectively [124, 125].
Although involvement of ANT in the MPTP formation was proposed in 1990 and subsequent evidence supporting its role in mitochondrial permeable transition has been considered more convincing than in case of VDAC, its role as channel-forming subunit of the MPTP has recently been questioned [96, 107, 126, 127]. Specific inhibitors of ANT affect Ca2+-induced mitochondrial permeabilization and apoptosis [107, 128–130]. ANT1, but not ANT2, has been demonstrated to be associated with CypD at contact sites where the MPTP is predominantly located [131]. Moreover, reconstitution of an ANT–CypD complex in liposomes has resulted in formation of a channel resembling the MPTP [115, 116]. Finally, several laboratories have demonstrated that overexpression of ANT1, but not ANT2, which does not interact with CypD, leads to mitochondrial cell death in various cultured cells including cardiomyocytes [132–134].
While all these findings suggest that ANT is indeed an essential subunit of the MPTP, as with VDAC, data from transgenic mice contradict this conclusion. Surprisingly, ANT1/2 double knockout mice still exhibit a cyclosporine A (CsA)-sensitive MPTP; however, significantly higher Ca2+ concentrations are required to induce MPTP opening [135]. In addition, MPTP opening in the ANT-deficient mitochondria has been completely insensitive to adenine nucleotides and ANT ligands [135]. Although the presence of a novel ANT4 isoform in mouse mitochondria further complicates the situation, according to current consensus ANT is not a bona fide pore-forming component of the MPTP but rather an auxiliary regulatory subunit responsible for the pore sensitivity to adenine nucleotide, ANT ligands, and, to a certain extent, Ca2+ [136].
Cyclophilin-D
Demonstration that Ca2+-induced MPTP opening can be inhibited by sub-micromolar concentrations of CsA has been pivotal in our understanding of the MPTP functioning [137]. The CsA target has subsequently been identified as a novel mitochondrial matrix cyclophilin, CypD [108, 138]. Furthermore, Ca2+ has inhibited the CsA binding to CypD, implying that CypD could be involved in Ca2+ regulation of the MPTP. CypD, a peptidylprolyl isomerase, is encoded by the nuclear PPIF gene, synthesized in the cytosol, and targeted to mitochondria due to a mitochondrial targeting sequence [139, 140]. CypD isomerase activity is essential for MPTP opening since its inhibition has resulted in attenuation of mitochondrial permeabilization and cell death in various cells [141–144]. However, it should be noted that CypD is a small (∼18 kDa) soluble matrix protein, and therefore it cannot be the pore-forming component.
In contrast to VDAC and ANT, genetic approach has confirmed a critical role of CypD as a component of the MPTP: Ppif −/− mice are resistant to Ca2+– and OS-induced mitochondrial permeabilization and cell death [145–148]. Significantly, expression of functional CypD has resensitized the Ppif −/− cells to mitochondrial permeabilization and cell death, while an isomerase-inactive mutant of CypD has failed to rescue the phenotype [145]. However, the loss of CypD does not attenuate mitochondrial permeabilization completely: it rather significantly reduces sensitivity of the MPTP to Ca2+-overload and OS.
The CypD-deficient mice have been extensively studied with regard to myocardial disorders. It has been reported that loss of CypD results in significantly higher resistance to myocardial I/R injury compared to wild type animals [145, 146]. Consistently, in the CypD-deficient mice pre- and postconditioning have not resulted in protection against I/R injury [149]. Moreover, the Ppif −/− mice have been reported to show reduced mitochondrial-dependent necrosis associated with cardiomyopathy [104, 150]. These data suggest that targeting CypD inhibits the MPTP and thereby represents an essential cardioprotective mechanism.
Mitochondrial Phosphate Carrier (PiC)
Three other members of the SLC25 family have the potential to create a pore-like structure in the MIM: the aspartate–glutamate carrier (AGC), the ornithine–citrulline carrier (ORC), and the mitochondrial phosphate carrier (PiC) [95, 151]. Two AGC isoforms, encoded by the SLC25a12 and SLC25a13 genes, mediate the import of glutamate and H+ into the mitochondria in exchange for aspartate, while the ORC, encoded by the SLC25a15 gene, transports ornithine into the mitochondria in exchange for citrulline and H+ [125]. Although there are data, which suggest that the AGC and ORC may act as components of the MPTP, their potential roles remain to be determined [96, 152–154].
Given the ability of inorganic phosphate (Pi) to sensitize the MPTP opening induced by Ca2+ and ROS, the mitochondrial PiC represents an obvious candidate for a pore-forming subunit of the MPTP. The PiC is a ubiquitously expressed protein of ∼30 kDa, located in the MIM; it is encoded by the SLC25a3 gene and the gene product is processed to generate two splice variants [125]. The PiC promotes the Pi reuptake into the mitochondrial matrix. It has recently been demonstrated that the PiC interacts with CypD and this interaction has been enhanced by MPTP-inducing agents and reduced by MPTP blockers [155]. The PiC also associates with ANT contributing to the formation of the mitochondrial ATP synthasome [156]. Moreover, reconstitution of the PiC in lipid vesicles can lead to formation of a nonspecific pore [157]. Finally, a high-throughput screen for cell death inducers has recently identified that overexpression of the PiC initiates cytochrome c release and apoptosis [158]. Although all these data suggest that the PiC can be a component of the MPTP, it remains to be determined if the PiC is a bona fide pore-forming subunit of the MPTP, or it is another regulatory subunit, like ANT or CypD (Fig. 3.5b). Rigorous genetic analyses using gene- or siRNA-knockdown approach are required to unambiguously address the role of PiC in the MPTP.
It has been suggested that the ETC machinery, complex I in particular, can not only regulate directly the MPTP but be the pore itself [159]. However, the majority of available data do not support this hypothesis and suggest that complex I indirectly regulates the MPTP affecting the levels of mitochondria-derived ROS [96]. Lemasters has suggested that any misfolded protein of the MIM, produced during cellular stress, can act as a pore, while the CypD regulator binds to such misfolded protein due to its chaperone-like capacity [160]. Nevertheless, according to the current model, VDAC is no longer a component of the MPTP, PiC is added to the model as a candidate for the pore-forming subunit, while ANT and CypD represent regulatory subunits of the MPTP (Fig. 3.5b).
Mitochondrial Channels
Mitochondria communicate with the cell via numerous channels. K+, Na+, Ca2+, H+ as well as Pi, nucleotides, and various proteins cross the MOM and MIM in a highly regulated manner to orchestrate a variety of cellular processes ranging from ATP synthesis and mitochondria dynamic organization to cell survival and death. Historically, VDAC was the first channel identified in mitochondria, quickly followed by the first characterization of MPTP [98, 161]. A comparison of their channel activities is summarized in Table 3.1. Since then great progress has been made in our understanding of the molecular structure and mechanism of various mitochondrial channels and their critical roles in the heart physiology and pathophysiology.
Table 3.1
Main channel features of VDAC and MPTP
Parameter | VDAC | MPTP |
|---|---|---|
Location | MOM | MIM |
Pore diameter (nm) | 2.2 ± 0.05 | 2.8 ± 0.1 |
Voltage dependence | Yes | Yes |
Ion selectivity | Anion | Slightly cation |
P K+/P Cl− | 0.7 ± 0.1 | 7.0 |
Physiological modulators | NADH | ADP, Ca2+, Pi |
Pharmacologic modulators | DIDS, RuR, Ru360 | CsA |
Mitochondrial Ca2+ Channels
Ca2+ transport across the mitochondrial membranes, mediated by mitochondrial Ca2+ channels, plays critical roles in a variety of cellular signaling pathways regulating a myriad of myocardial physiological and pathophysiological processes [162–165]. Ca2+ uptake through mitochondrial Ca2+ channels regulates ATP production and thereby allows mitochondria to react on changes in cardiomyocyte energy needs [166]. However, mitochondrial Ca2+ overload can induce the generation of ROS and MPTP opening leading eventually to cell death (Fig. 3.6) [127, 162, 167, 168].
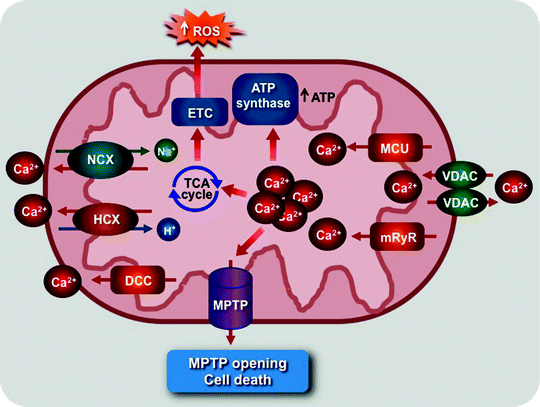
Fig. 3.6
Scheme of role of mitochondrial Ca2+ channel and transporters. The mitochondrial Ca2+ uniporter (MCU) and ryanodine receptor (mRyR) mediate mitochondrial Ca2+ uptake, which stimulates the tricarboxylic acid (TCA) cycle, elevating NADH/FADH2 production to feed electrons via the electron transport chain (ETC), and activates the ATP synthase. Mitochondrial Ca2+ overload can also activate reactive oxygen species (ROS) generation triggering mitochondrial permeability transition pore (MPTP) opening and cell death. The MPTP, voltage-dependent anion channel (VDAC), mitochondrial H+/Ca2+ (HCX), and Na+/Ca2+ (NCX) exchangers and diacylglycerol-activated cation channels (DCC) mediate Ca2+ efflux
Mitochondrial Ca2+ uptake was originally considered to be mediated by the mitochondrial Ca2+ uniporter (MCU). Electrophysiological and biochemical studies have subsequently identified several seemingly different channels including besides MCU, MiCa, mCa1, mCa2, RaM, and the mitochondrial ryanodine receptor (mRyR) (Table 3.2) [166, 169–174]. The key common characteristic of these channels is their sensitivity to the specific inhibitor Ru360.
Table 3.2
Main biophysical characteristics of mitochondrial Ca2+ channels
Channel | Ion selectivity | Current | Molecular identity | Activators | Inhibitors |
|---|---|---|---|---|---|
MCU | |||||
MiCa | Highly Ca2+ selective | Inward rectifying | – | – | Ru360, RuR |
mCa1 | Highly Ca2+ selective | Inward rectifying | – | Spermine | Ru360, RuR |
mCa2 | Highly Ca2+ selective | inward rectifying | – | Spermine | relatively insensitive to Ru360 |
VDAC | Anion-/cation-selective state, PCa2+/PCl− ∼0.02–0.38 | Closed at >±40 mV | VDAC 1,2,3 | – | DIDS, RuR, Ru360 |
mRyR | Cation selective, PCa2+/PK+ ∼6/1 | Linear | RyR1 | <10 μM ryanodine, impera toxin A, spermine | >100 μM ryanodine |
DCC | Slightly cation selective, Ca2+ selectivity undefined | Linear | – | DAG | 1 mM La2+ |
VDAC mediates Ca2+ and metabolite (e.g., ATP and ADP) transport across the MOM into the IMS [175, 176]. Although the MOM has initially been considered not to be a barrier for Ca2+, VDAC overexpression enhances the Ca2+ transport to mitochondria suggesting that the MOM does limit mitochondrial Ca2+ uptake [177]. Ca2+ can also control the permeability of the MOM: it induces a prolonged open state of VDAC increasing thereby metabolite transport [178].
The Ca2+ levels inside mitochondria are also controlled by several Ca2+ efflux pathways (Fig. 3.6). A second messenger, diacylglycerol (DAG), activates the novel DAG-activated cation channel (DCC), located in the MIM, to release Ca2+ from the Ca2+-loaded mitochondria [179]. Mitochondrial Na+/Ca2+ and H+/Ca2+ exchangers can mediate either Ca2+ uptake or release depending on the concentration of Na+, H+, and Ca2+ [164]. However, the biophysical characteristics of these exchangers remain to be determined. Moreover, mitochondrial Ca2+ efflux occurs during MPTP opening [127, 162].
Despite intensive efforts over the years, only the molecular identity of mRyR has been identified, while the identities of the remaining mitochondrial Ca2+ channels are elusive, and they have even been suggested to be different kinetic states of MCU [165]. A 500-kDa ryanodine receptor was first identified in the MIM of rat heart mitochondria in 2001 [173]. Cardiac mRyR is related to the skeletal muscle type 1 isoform (RyR1) rather than to cardiac RyR2, which is located in the SR [166]. Kinetic characteristics of mRyR imply that it acts specifically to optimize Ca2+ transport between mitochondria and the SR in the myocardium.
Thus, mitochondrial Ca2+ transport is orchestrated by a fine-tuned interplay between multiple Ca2+ channels and exchangers located in the MOM and MIM. Dysfunction of these mechanisms contributes to various cardiac diseases.
Mitochondrial K+ Channels
K+ represents the most abundant intracellular cation; its concentrations inside the mitochondria are approximately 150 mM similar to those in the cytosol [180]. Under physiological conditions, mitochondrial K+ channels mediate K+ influx into mitochondria, while the K+/H+ exchanger mediates K+ efflux in exchange for protons (Fig. 3.7). K+ influx into the mitochondrial matrix is accompanied by counter anion (mainly Pi) uptake and water diffusion and therefore regulates mitochondrial volume affecting structure and function [180, 181].
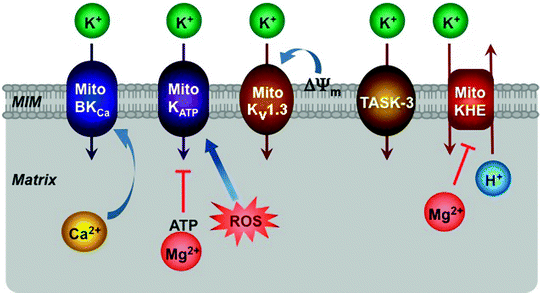
Fig. 3.7
Schematic representation of K+ channels located within the mitochondrial inner membrane (MIM). ATP-regulated (MitoKATP), large-conductance Ca2+-activated (MitoBKCa), voltage-dependent (MitoKv1.3), twin-pore (TASK-3) K+ channels and K+/H+ exchanger (MitoKHE) along with their major effectors are depicted. See text for the details. Most of these interactions occur within mitochondrial membrane; however, in this simplified scheme its important role is not shown. ROS reactive oxygen species, ΔΨ m mitochondrial membrane potential
Several mitochondrial K+ channels, located in the MIM, have been described, including the ATP-regulated K+ channel (mitoKATP), the large conductance Ca2+-activated K+ channel (mitoBKCa), the voltage-gated K+ channel (mitoKV1.3), and the twin-pore domain TASK-3 K+ channel (Fig. 3.7) [182–185]. Some mitochondrial K+ channels appear to resemble their counterparts present in the plasma membrane; however, the molecular identities of these channels remain yet elusive.
mitoKATP Channel
ATP- and glibenclamide-sensitive small conductance K+ current was described in beef heart mitochondria almost 20 years ago [186]. The pharmacological characteristics and the immunoreactivity with specific antibodies suggest that the mitoKATP channel is a member of the inward-rectifier K+ channel family, Kir6.x [187, 188]. However, though a few candidates for the channel subunits have been identified using specific antibodies against Kir6.1, Kir6.2, and sulfonylurea regulatory subunit (SUR2A), subsequently it appears that the mitoKATP channel does not contain any of the traditional KATP channel subunits [186, 189–192].
mitoBKCa Channel
The mitoBKCa channel with characteristics similar to the plasma membrane Ca2+-activated K+ channel was indentified in the mitoplasts of guinea pig ventricular cells by O’Rourke’s laboratory [193]. Similar to its plasma membrane counterpart, the mitoBKCa channel can be activated by NS1619 and inhibited by charybdotoxin. Importantly, its activation with NS1619, which leads to mitochondrial depolarization and inhibition OXPHOS, reduces infarction size after myocardial I/R [193–195]. Interaction between the mitoBKCa channel and cytochrome c oxidase-1, which can affect mitochondrial ATP and/or ROS production, may underlie the cardioprotective effect of K+ influx into mitochondria [196]. In contrast to the mitoKATP channel, the mitoBKCa channel appears to be not only functionally but also structurally similar to its surface membrane counterpart. Putative candidates for α- and β-subunits of the mitoBKCa channel have been reported [197–199].
mitoKV1.3 Channel
The presence of the mitoKV1.3 channel, with electrophysiological characteristics similar to its plasma membrane counterpart, has been found in mitoplasts isolated from T lymphocytes [184]. Inhibition of the mitoKV1.3 channel results in mitochondrial hyperpolarization. Importantly, pro-apoptotic Bax can interact with and inhibit the channel, and consistently KV1.3-deficient lymphocytes are resistant to Bax-induced apoptosis [200]. Thus, in contrast to the cardioprotective effect of activation of the mitoKATP and mitoBKCa channels, the mitoKV1.3 channel opening appears to mediate cell death at least in lymphocytes. It remains to be determined if the mitoKV1.3 channel plays pro-apoptotic role also in cardiomyocytes.
Finally, using molecular biology and immunological methods one of the twin-pore domain K+ channels, TASK-3, has recently been found in mitochondria of melanoma and keratinocyte cells [185]. However, the molecular identity and functional characteristics of this channel are currently unknown.
Mitochondrial Protein Import Channels
Human mitochondrial genome encodes only 13 of the 1,500 different mitochondrial proteins. Therefore, the vast majority of mitochondrial proteins is synthesized on cytosolic ribosomes and must be transported to their intramitochondrial destination. To pass through one or both mitochondrial membranes, precursor proteins should contain mitochondrial targeting signals.
Cleavable presequences represent the classical signals; they have a length of usually 15–50 amino acid residues, located at the N-terminus of precursor proteins, and form positively charged amphipathic α-helices. Most presequences are cleaved off by the mitochondrial processing peptidase (MPP) in the matrix, while some hydrophobic signals are removed by the MIM peptidase. All MOM proteins, most IMS proteins, many multispanning MIM proteins, and a few matrix proteins contain noncleavable internal targeting signals, located at various positions within their molecules [201]. The exact targeting mechanisms of these internal signals are currently unknown.
Gathered evidence suggests the existence of various complex pathways for the importing and sorting of mitochondrial proteins, they will be overviewed in Chap. 4. In this section, mainly the structural aspects of the outer and inner membrane translocases, TOM and TIM, respectively, will be discussed.
TOM
TOM complex represents the general portal and almost all mitochondrial precursor proteins enter through this gate. Tom40 is the central, pore-forming component of TOM complex (Fig. 3.8). This integral membrane protein has a β-barrel structure, which creates the channel for preprotein transport across the MOM. Tom40 forms oligomers yielding two to three protein-conducting channels per TOM complex [203, 204]. The peptide-sensitive channel activity of TOM was first demonstrated in MOM preparations in 1997 and subsequently was attributed to Tom40 [205, 206]. TOM channel activity is sensitive to peptides harboring target sequences; such peptides induce a rapid flickering (up to 500 transitions/sec) of the channel.
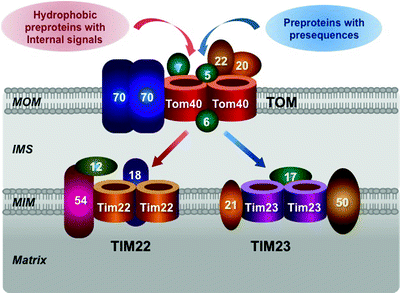
Fig. 3.8
Molecular structure of mitochondrial protein import apparatus. Major components of the translocase of the MOM (TOM) complex, located in the mitochondrial outer membrane (MOM), and the translocase of the MIM (TIM) 22 and TIM23, located in the mitochondrial inner membrane (MIM), are schematically shown. See text for details. IMS intermembrane space
The TOM complex contains three receptor subunits Tom20, Tom22, and Tom70. Tom20 recognizes precursor proteins with presequences, while Tom70 recognizes preproteins with various internal targeting signals, e.g., precursors of MIM metabolite carriers [207–209]. Tom20 and Tom70 transfer corresponding preproteins to Tom22, which inserts them into the protein-conducting channel formed by Tom40 (Fig. 3.8).
Tom22 acts not only as the central receptor subunit but also as an organizer of the 450 kDa TOM complex. Loss of Tom22 results in dissociation of the complex into smaller Tom40-containing subcomplexes [209–211]. The small auxiliary subunits Tom5, Tom6, and Tom7 also contribute to assembly and stability of the TOM complex [210, 212, 213]. Tom5 is involved not only in the formation of the TOM complex but also in the delivery of preproteins from Tom22 to the Tom40 channel. Tom6 mediates the 450 kDa TOM complex stability, while Tom7 facilitates its dissociation. Finally, the imported proteins are transferred to TIM22 or TIM23 to be targeted to their final locations [201, 214].
Presequence Translocase in the MIM (TIM23)
Transport of preproteins with presequences across the MIM into the mitochondrial matrix is mediated by two highly complex machineries: TIM23 complex and its associated matrix-exposed import motor, PAM. TIM23-PAM functions in cooperation with the TOM complex as well as with OXPHOS, using two energy sources: ATP and the electrochemical mitochondrial membrane potential (ΔΨm).
The core components of the TIM23 complex are the channel-forming Tim23, the receptor Tim50, and Tim17 involved in PAM recruitment and lateral preprotein sorting [215–220]. Tim50, Tim23, and Tim21 communicate with the TOM complex promoting translocation of precursor proteins from the MOM to the MIM [217–219]. In addition, Tim21 is involved in the coupling of the TIM23 complex with the ETC complexes III and IV, which provide energy for the ΔΨm-driven TIM23 machinery [221–223]. ΔΨm activates the channel-forming Tim23 and, being negative on the matrix side, facilitates translocation of the positively charged presequences of preproteins [215, 224].
The molecular chaperone heat shock protein 70 (mtHsp70) plays a key role in the ATP-consuming motor PAM. mtHsp70 binds to and translocates preproteins into the matrix. This process requires ATP hydrolysis. Mge1, a nucleotide exchange factor, stimulates ATP cleavage cycle by releasing ADP from mtHsp70. Four other components of PAM are membrane-bound cochaperones involved in the TIM23-PAM interaction. Tim44 serves as an ATP-dependent binding site for mtHsp70 close to the Tim23 channel; it also binds to the preprotein emerging on the matrix side of the channel [224, 225]. J-domain-related chaperones Pam16 and Pam18 form a stable complex, which activates the ATPase activity of mtHsp70 [226, 227]. Moreover, Pam17 binds to Tim23, while Pam18 binds to Tim17 [219, 228, 229].
Carrier Translocase in the MIM (TIM22)
Cytosolic chaperones Hsp70 and Hsp90 bind the hydrophobic precursor carrier proteins, such as PiC, synthesized on cytosolic ribosomes. The receptor Tom70, containing binding sites for both the preproteins and the chaperones, recognizes and binds these complexes [230–233] (Fig. 3.8). The chaperone–preprotein complex dissociates in an ATP-dependent manner, and the preprotein is translocated into the Tom40 channel [230].
In the IMS, the hexameric Tim9–Tim10 complex binds the hydrophobic carrier preproteins and transported them to the TIM22 complex [234–237] (Fig. 3.8). Soluble Tim9–Tim10 complex binds Tim12 to form a membrane-bound Tim9–Tim10–Tim12 complex at the outer surface of the TIM22 complex [214, 238]. Tim54, a membrane-embedded subunit of the TIM22 complex, may serve as a binding site for the Tim9–Tim10–Tim12 complex [239, 240]. Thus, carrier precursor proteins are delivered to the translocase channel formed by Tim22 [238, 241]. The TIM22 complex is a twin-pore translocase driven by ΔΨm [238]. Finally, in the MIM, carrier precursor proteins are released and assembled into their mature forms; however, the mechanism of these steps remains to be determined.
Mitochondrial Matrix and Intermembrane Space
The mitochondrial matrix compartment enclosed by the inner membrane contains a compact genome, circular, double-stranded mitochondrial DNA (mtDNA), RNAs along with enzyme machineries involved in mitochondrial nucleic acids transactions, as well as their own ribosomes (it will be discussed in detail in Chap. 4). The mitochondrial matrix is highly enriched in proteins, containing approximately two-thirds of the total mitochondrial protein [40]. Matrix proteins are implicated in a variety of essential processes, ranging from OXPHOS, the Krebs cycle, and fatty acid oxidation (FAO) to transport of substrates, ions, and nucleotides through mitochondrial membranes, signal transduction, and apoptosis.
The IMS, wrapped by the MOM and MIM, contains much lower protein amounts (approximately 6% of the total mitochondrial protein) compared to the matrix [40]. Proteins of the IMS have the specific leader or targeting amino acid sequences, which are required to cross the mitochondrial membrane. Among the major IMS proteins are several pro-apoptotic factors including cytochrome c, Smac/DIABLO, endonuclease-G and AIF [103].
In addition, the IMS contains also two metabolic kinases: mitochondrial creatine kinase (MtCK) and nucleotide diphosphate kinase (NDPK-D). Both kinases contain canonical mitochondrial import presequences and interact with a high affinity with anionic phospholipids, with CL in particular [242–244]. MtCK catalyzes the reversible phosphorylation of creatine to generate high-energy phosphate pools in the form of phosphocreatine, while NDPK-D promotes exchange of the γ-phosphate between tri- and diphosphonucleosides [79, 245–247]. Surprisingly, these two phylogenetically unrelated kinases share functionally essential properties: they maintain proper nucleotide pools critical for cellular energy homeostasis, interact with ANT through high affinity binding to CL and contribute to the intermembrane lipid transfer as well as to apoptotic response.
Mitochondrial Dynamics
In the past two decades, the concept of mitochondria as relatively static organelles has significantly changed. It has been demonstrated that mitochondria can move along cytoskeletal tracks and modify their morphology, intracellular distribution, and activity by constant highly regulated fusion and fission events. These highly regulated processes allow the cell to respond properly to frequently changing conditions. A shift toward fusion results in the formation of large mitochondrial networks, found in metabolically active cells, while a shift toward fission leads to mitochondrial fragmentation, observed in quiescent cells [248, 249]. Moreover, the elongation of mitochondrial tubules has been shown upon differentiation of embryonic stem cells into cardiomyocytes, while mitochondrial fragmentation can contribute to cytochrome c release and eventually to apoptosis [250, 251]. Evolutionary-conserved large GTPases, related to the dynamin family proteins, along with their interacting proteins constitute the core machinery, which controls mitochondrial dynamics (Table 3.3) [252, 253].
Table 3.3
Core components of the mitochondrial fusion and fission machineries
Human protein | Location | Proposed function |
|---|---|---|
Fusion machinery | ||
MFN1/MFN2 | MOM | MOM fusion |
OPA1 | MIM (l-OPA1) MIM and IMS (s-OPA1) | MIM fusion |
Fission machinery | ||
DRP1 (also known as DLP1) | MOM and cytosol | MOM fission |
FIS1 | MOM | Putative factor for DRP1 recruitment |
MFF | MOM | Putative alternative factor for DRP1 recruitment |
The Molecular Machinery and Mechanisms of Mitochondrial Fusion
Fusion of the MOM is mediated by members of mitofusin family conserved in yeast, worms, flies, and mammals [254–258]. Mitofusins are large GTPases, which share a similar molecular architecture: two transmembrane domains, which anchor the proteins in the MOM, a short loop in the IMS and the C-terminal regions protruding into the cytosol (Fig. 3.9) [259, 260]. In mammals, there are two mitofusin isoforms, MFN1 and MFN2. Both MFN1 and MFN2 are localized in the MOM and can interact with their counterparts in adjacent mitochondria to coordinate mitochondrial fusion [260, 261].
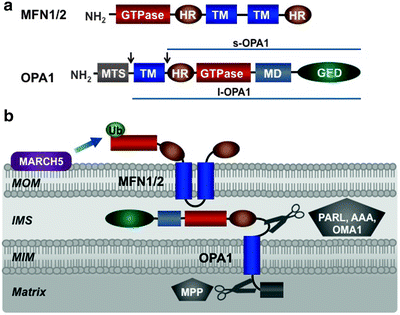
Fig. 3.9
Mitochondrial fusion machinery. (a) Molecular structure of mitofusin (MFN) 1/2 and optic atrophy protein (OPA) 1. MFN1/2 is located in the mitochondrial outer membrane (MOM) and comprises a GTPase domain, two heptad-repeat regions (HR), and two transmembrane domains (TM). OPA1 is located in the mitochondrial inner membrane (MIM) and contains mitochondrial targeting sequence (MTS), transmembrane domain (TM), heptad-repeat region (HR), GTPase domain, middle domain (MD), and GTPase effector domain (GED). Alternative splicing and processing of OPA1, shown by arrows, generate long (l-OPA1) and short (s-OPA1) isoforms. (b) Assembly and posttranslational modifications of mitochondrial fusion machinery within mitochondrial membranes. See text for details. AAA mitochondrial protease “ATPase associated with diverse cellular activities,” IMS intermembrane space, MARCH5 ubiquitin-protein ligase “membrane-associated ring finger (C3HC4) 5,” MPP matrix processing peptidase, OMA mitochondrial protease “overlapping activity with m-AAA protease,” PARL mitochondrial protease “presenilins-associated rhomboid-like,” Ub ubiquitin
Mammalian optic atrophy protein 1 (OPA1) mediates fusion of the MIM [262]. This is a dynamin-related large GTPase, and related proteins have been identified in yeast, worms, and flies [256, 263, 264]. OPA1 has an N-terminal mitochondrial targeting sequence cleaved by matrix-processing peptidase (MPP), a GTPase domain, a GTPase effector domain, and multiple heptad repeats (Fig. 3.9a). Eight OPA1 isoforms result from alternative splicing and alternative processing at two sites between the N-terminal transmembrane region and heptad repeats [265, 266]. In mammals, alternative processing of OPA1 is catalyzed by the presenilins-associated rhomboid-like protease (PARL), the IMS and matrix AAA proteases, and the MIM peptidase OMA1 (also referred to as MPRP1) [267–271]. As a result, long OPA1 isoforms (l-OPA1), containing an N-terminal transmembrane domain, and short isoforms (s-OPA1), lacking the transmembrane domain, can be generated (Fig. 3.9a). Both long and short isoforms have a GTPase domain, a GTPase effector domain and multiple heptad repeats. L-OPA1 is anchored to the MIM by its transmembrane domain, while s-OPA is targeted to the MIM via its association with the MIM-anchored l-OPA1 [267, 268]. A fraction of s-OPA1 is believed to communicate with mitofusins in the MOM.
Mammalian MFN1 or MFN2 together with OPA1 represents the core component of the mitochondrial fusion machinery (Fig. 3.9b). Consistent with their critical roles in fusion, their deficiencies lead to fragmentation of tubular mitochondria network [261, 272, 273]. Importantly, reexpression of OPA1 has restored interconnected mitochondrial tubules [262].
According to the current model of mitochondrial fusion, the C-terminal heptad repeats of mitofusins of adjacent mitochondria interact to form an intermolecular antiparallel coiled coil to tether approaching mitochondria [274]. This initial docking step brings the MOM of two mitochondria close together to initiate phospholipid mixing. The GTPase activity of MFN1 or MFN2 provides energy for these processes. L-OPA1 appears to mediate tethering of opposing MIM, while the GTPase activity of s-OPA1, which is activated upon binding to l-OPA1, generates energy required for lipid bilayer mixing and fusion of the MIM [275].
Although fusion of the MOM and MIM is mediated by distinct machineries, under normal conditions, these processes should occur in a highly coordinated fashion to maintain mitochondria integrity. In yeast, the MOM protein Ugo1 coordinates the MOM and MIM fusion, physically interacting with both Fzo1 and Mgm1, yeast orthologues of MFN1 and OPA1, respectively [276, 277]. However, no structural or functional Ugo1 equivalents in higher eukaryotes have been found. Thus, the molecular mechanisms underlying the coupling of the MOM and MIM fusion remain to be determined.
The Molecular Machinery and Mechanisms of Mitochondrial Fission
In mammals, a dynamin-related protein 1 (DRP1, also known as DLP1; yeast orthologue Dnm1) and mitochondrial fission protein 1 (FIS1; yeast orthologue Fis1) are key components of the mitochondrial fission machinery. DRP1 contains an N-terminal GTPase domain, middle domain, and a C-terminal GTPase effector domain essential for self-assembly (Fig. 3.10a). DRP1 is a soluble cytosolic protein but its subpool colocalizes with mitochondria at sites of future fission [278, 279].
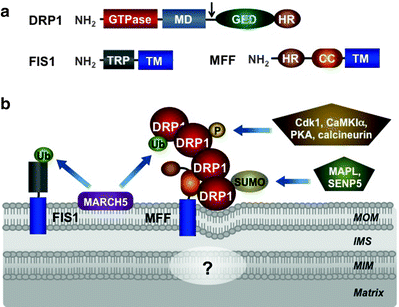
Fig. 3.10
Mitochondrial fission machinery. (a) Molecular structure of dynamin-related protein 1 (DRP1), mitochondrial fission protein 1 (FIS1), and mitochondrial fission factor (MFF). DRP1 is predominantly located in the cytosol and contains a GTPase domain, middle domain (MD), GTPase effector domain (GED), and heptad-repeat region (HR). Alternative splicing site is shown by arrow. FIS1 and MFF are anchored by their transmembrane domains (TMs) in the mitochondrial outer membrane (MOM). In addition, FIS1 contains also tetratricopeptide repeat (TRP), while MFF contains heptad-repeat region (HR), and coiled-coil (CC) region. (b) Assembly and posttranslational modifications of mitochondrial fission machinery. According to the current model, MFF acts as a central recruiter of DRP1 to the mitochondrial surface. Recruited DRP1 forms spiral structures around the mitochondria resulting eventually in mitochondrial membrane constrictions and division. FIS1 and additional factors may contribute to the assembly of the fission apparatus. Currently, it is very little known about mechanisms of the mitochondrial inner membrane (MIM) division. See text for details. Other abbreviations: CaMK1α calcium/calmodulin-dependent protein kinase 1 alpha, Cdk1 cyclin-dependent kinase 1, IMS intermembrane space, MAPL mitochondria-anchored protein ligase, MARCH5 ubiquitin-protein ligase “membrane-associated ring finger (C3HC4) 5,” SENP5 sentrin-specific protease 5, SUMO small ubiquitin-like modifier protein, Ub ubiquitin
FIS1 is a single-pass transmembrane protein with a C-tail anchored in the MOM [280–282]. Its multiple tetratricopeptide repeat motifs, facing the cytosol, are thought to be involved in the recruitment of DRP1 to mitochondria (Fig. 3.10a) [283–285]. Although previous studies have demonstrated that overexpression of FIS1 has led to mitochondrial fragmentation and its depletion has resulted in interconnected mitochondrial networks caution should be taken interpreting these data, as these manipulations can induce nonphysiological stress causing perturbations of the mitochondrial morphology [281, 282]. Human FIS1 localizes uniformly on the MOM, in contrast to the punctuate DRP1 foci, and its depletion does not affect mitochondrial recruitment of DRP1 or mitochondrial morphology [286, 287]. Consistently, knockout of two homologous FIS1 genes in Caenorhabditis elegans does not cause any detectable mitochondrial fission phenotype [288]. Thus, while mammalian FIS1 can interact with DRP1, its actual function remains unclear, and alternative pathways of the mitochondrial DRP1 recruitment may exist [286, 288, 289].
Mitochondrial fission factor (MFF) may play a role of such an alternative fission factor. MFF has N-terminal heptad repeats, coiled-coil domain, and a C-terminal transmembrane tail anchored in the MOM (Fig. 3.10a) [290]. MFF and FIS1 are present in separate 200-kDa complexes, suggesting that they mediate different pathways of mitochondrial fission. Consistent with its role in mitochondrial fission, silencing of MFF by RNAi inhibits mitochondrial and peroxisomal fission and delays cytochrome c release and further progression of apoptosis [290]. Importantly, the mitochondrial recruitment of DRP1 has been decreased in MFF RNAi cells, while MFF overexpression has induced DRP1 recruitment to mitochondria concomitant with mitochondrial fragmentation [287]. Moreover, in contrast to the uniform localization of FIS1 in the MOM, MFF mainly colocalizes with the DRP1 foci on the MOM. Thus, MFF is restricted to metazoans, and mechanisms of the mitochondrial fission in metazoans are different from those of yeast and plant.
According to the current model, MFF acts as a central recruiter of DRP1 to the mitochondrial surface (Fig. 3.10b). Recruited DRP1, due to its ability to homo-oligomerize, forms spiral structures around the mitochondrial tubules. FIS1 and additional factors may contribute to the assembly of the fission apparatus resulting eventually in mitochondrial membrane constrictions and division [291, 292].
Ganglioside-induced differentiation-associated protein 1 (GDAP1) and endophilin B1 (Endo B1) may represent such additional factors, involved in the mitochondrial fission. GDAP1 is a single-pass transmembrane protein anchored by its C-tail in the MOM [293, 294]. The loss of GDAP1 results in elongated mitochondria, while its overexpression induces DRP1-mediated mitochondrial fragmentation. Moreover, mutations in the GDAP1 gene cause the peripheral neuropathy Charcot–Marie–Tooth, and mutated GDAP1 identified in these patients has no or significantly reduced fission activity [293]. Like DRP1, Endo B1 can be recruited to the MOM accompanied by mitochondrial fission, and its silencing by RNAi affects mitochondrial morphology [295]. Although these data suggest that GDAP1 and Endo B1 contribute to the mitochondrial fission machinery, the exact molecular mechanisms of their actions remain to be determined.
It is conceivable that a distinct machinery for division of the MIM exists and functions in highly coordinated manner with the fission machinery in the MOM; however, very little is currently known about mechanisms of the MIM division. Mitochondrial protein of 18 kDa MTP18, located in the MIM, has been suggested to play a role in the MIM division. Depletion of MTP18 leads to mitochondrial tubules, while its overexpression results in mitochondrial fragmentation [296]. However, the mechanisms underlying these effects remain unknown.
Mitochondrial Trafficking
In addition to changes in morphology, mitochondria undergo constant changes in their intracellular location. Mitochondria are translocated along the microtubule network to subcellular compartments where the energy demands are high and/or where Ca2+ buffering is critical.
In humans, two mitochondrial Rho GTPases, MIRO1 and MIRO2, belonging to the Ras superfamily, are core components of the mitochondrial transport machinery [297–300]. The C-terminal transmembrane domains of the MIRO proteins are anchored in the MOM, while their two GTPase domains flanking two EF-hand motifs are exposed to the cytosol [301, 302]. The Rho GTPases are well-known regulators of cytoskeleton structures; however, the MIRO GTPases function as Ca2+ sensors in the microtubule-based mitochondrial transport complex. MIRO EF-hand domains bind Ca2+ and their deficiency attenuates cellular ability to cope with elevated intracellular Ca2+ levels [303–305]. Overexpression of wild-type MIROs, their constitutively active variants, and MIRO with mutated EF-hand domains have led to increased mitochondrial trafficking. Importantly, upon elevation of intracellular Ca2+ levels, wild-type MIRO and the constitutively active variants have inhibited mitochondrial motility, while MIRO with mutated EF-hands has been less sensitive to the increase in intracellular Ca2+ [303].
In mammals, kinesin 1 (KIF5B) and kinesin 3 (KIF1B) hydrolyze ATP to translocate mitochondria along microtubules toward their plus end from the cell body to periphery (anterograde direction) [306, 307]. The retrograde transport of mitochondria to the cell body is mediated by the microtubule-associated minus-end dynein motor [308, 309]. The coiled-coil domain proteins GRIF-1 (Drosophila orthologue Milton) and OIP106 (also known as TRAK2 and TRAK1, respectively) act as adaptors, which facilitate interactions between kinesin motors and MIROs [302, 310]. The GTPase activity of MIRO1 is essential for the recruitment of GRIF-1 and regulates thereby mitochondrial motility [311].
Ca2+ as a universal signaling molecule is involved in the control of various energy-consuming processes through the recruitment of mitochondria to subcellular compartments of elevated Ca2+ levels. Maximal axonal movement of mitochondria has been observed at resting Ca2+ concentrations (<0.1 mM), whereas increase in intracellular Ca2+ inhibited mitochondrial motility [312–314]. Recently, two models of mitochondrial trafficking in response to intracellular Ca2+ fluctuations have been proposed. Both models are based on the central role of the MIRO EF-hand motifs as Ca2+ sensors in the microtubule-based mitochondrial transport machinery (Fig. 3.11).
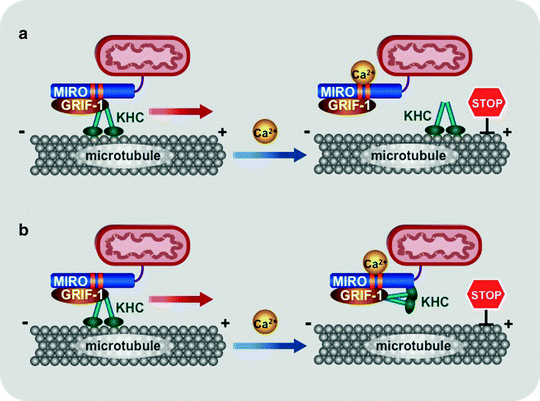
Fig. 3.11
Scheme of two models for mitochondrial trafficking. At resting Ca2+ concentrations (<0.1 mM), the kinesin-GRIF-1/Milton-MIRO complex are assembled and translocates mitochondria along microtubules. (a) The first model proposes that increased Ca2+ levels induce dissociation of GRIF-1-MIRO from kinesin (KHC). (b) According to the second model, elevated Ca2+ concentrations change the binding preferences of kinesin motor domain from microtubules to MIRO. Regardless of the exact mechanism, high local Ca2+ concentrations inhibit mitochondrial movement, retaining mitochondria at areas, where ATP generation and/or Ca2+ buffering are mostly required. Abbreviations: GRIF-1 gamma-aminobutyric acid receptor-interacting factor, MIRO mitochondrial Rho-GTPase protein
In both models, at resting Ca2+ concentrations (<0.1 mM), the kinesin–GRIF-1/Milton–MIRO complex are assembled and mitochondria are translocated by this complex along microtubules. The first model suggests that increase in Ca2+ levels induces dissociation of GRIF-1–MIRO from kinesin (Fig. 3.11a) [304]. According to the second model, elevated Ca2+ concentrations change the binding preferences of kinesin motor domain from microtubules to MIRO (Fig. 3.11b) [305]. The KIF5 motor appears to mediate the anterograde mitochondrial transport, whereas the motor responsible for the retrograde movement is currently unknown. Regardless of the exact mechanism, high local Ca2+ concentrations induce arrest of both anterograde and retrograde mitochondrial movement, retaining mitochondria at areas where ATP generation and/or Ca2+ buffering are mostly required.
Regulation of Mitochondrial Dynamics
Antagonistic processes of mitochondrial fusion and fission are tightly regulated to maintain mitochondrial morphology under changing physiological conditions [315, 316]. Core components of mammalian machineries of mitochondrial dynamics, such as OPA1 and DRP1, represent the main targets for regulation.
Complex processing of OPA1, mediated by multiple proteases, such as PARL, OMA1, and several AAA proteases, generates various OPA1 splice variants (Fig. 3.9b) [266, 268–271]. Increased formation of s-OPA1 associated with mitochondrial fragmentation has been observed at low ATP levels, low ΔΨm and during apoptosis [268, 269, 317]. As low ΔΨm is a feature of damaged mitochondria and both l-OPA1 and s-OPA1 are required for mitochondrial fusion, elevated proteolytic conversion of OPA1 into s-OPA1 attenuates fusion of dysfunctional mitochondria [270, 271].
Ubiquitylation and sumoylation of key components of the core machineries of mitochondrial dynamics have recently emerged as an additional regulatory pathway. Posttranslational modifications are particularly important for regulation of DRP1. DRP1 overexpression does not affect mitochondrial fission suggesting that changes in its mitochondrial recruitment, GTPase activity, or self-assembly ability are more critical than its levels. The mitochondrial E3 ubiquitin-protein ligase MARCH5 (also known as MITOL), located in the MOM, binds and ubiquitylates MFN1/2, DRP1, and FIS1 (Figs. 3.9b and 3.10b) [318–321]. Importantly, deficiency of MARCH5 results in mitochondrial elongation and induces cellular senescence [321]. Mitochondrial-anchored protein ligase MAPL attaches small ubiquitin-like modifier (SUMO) to DRP1 and activates mitochondrial fission, while sentrin-specific protease 5 (SENP5) desumoylates DRP1 (Fig. 3.10b) [322, 323].
Emerging evidence suggests that PTEN-induced putative kinase 1 (PINK1) and parkin, encoded by two genes related to Parkinson’s disease, PINK1 and PARK2, respectively, may also be involved in the control of mitochondrial dynamics. PINK1 is a serine/threonine protein kinase located in the mitochondria, while parkin is a cytosolic E3 ubiquitin-protein ligase [324, 325]. Mitochondrial depolarization stabilizes PINK1 to recruit parkin to damaged mitochondria and direct them for mitophagy [325]. PINK1–parkin can mediate ubiquitination of the mitochondrial surface proteins VDAC and MFN2 [326, 327]. Currently, it is unclear whether these modifications affect the activity of these proteins or serve as targeting signals for mitophagy. It has been hypothesized that the parkin-mediated mono-ubiquitination of MNF2 inhibits fusion of damaged mitochondria with the mitochondrial network, while poly-ubiquitination of MNF2 induces mitophagy [327].
Three protein kinases, cyclin-dependent kinase 1 (CDK1)/cyclin B, cAMP-dependent protein kinase (PKA), and Ca2+/calmodulin-dependent protein kinase Iα (CaMKIα) phosphorylate various Ser residues of DRP1 modulating its fission activity [328–330]. The phosphatase calcineurin dephosphorylates DRP1 in a Ca2+-dependent manner contributing to the regulation of mitochondrial dynamics (Fig. 3.10b) [329, 331]. In addition, it has been reported that mitochondrial dynamics can be regulated transcriptionaly [332].
Recently, it has been demonstrated that PINK1 binds to the MIRO–OIP106 mitochondrial transport complex. Moreover, PINK1 depletion-induced fragmentation of mitochondria can be suppressed by overexpression of MIRO or Milton [333]. These findings link mitochondrial fusion and fission to mitochondrial trafficking adding thereby complexity to the regulation of mitochondrial dynamics.
In summary, highly regulated processes of mitochondrial dynamics play a critical role in the high energy demanding myocardium. A finely tuned balance between mitochondrial fusion and fission is essential for the maintenance of appropriate mitochondrial morphology and function. Growing evidence suggests that impaired mitochondrial dynamics contributes to the pathogenesis of severe neurodegenerative disorders and various cardiovascular diseases, including ischemia–reperfusion injury, cardiomyopathies, and heart failure. The role of mitochondrial dynamics in various human pathologies will be discussed in Chap. 17.
Conclusions
Since the 1950s, great advances in electron microscopy and tomography, the development of high-resolution fluorescent microscopy and an impressive progress in genomics and functional proteomics have revolutionized the field of mitochondria research. It had become evident that the mitochondria are not only powerhouses, which drives the constant pumping action of the myocardium, but central integrators of energy production, generation of ROS, various cell death pathways, and regulation of intracellular Ca2+ levels. Cardiac mitochondria are not viewed any more as static organelles since they undergo continuing changes in their intracellular locations and morphology. These highly dynamic mitochondrial rearrangements are mediated by complex multiprotein machineries of mitochondrial trafficking, fusion, and fission. Growing evidence suggests that these intricate processes play critical roles in cardiac function, while their impairments contribute to the pathogenesis of various cardiovascular disorders.
Despite the impressive progress in our understanding of mitochondrial structure and function, many important aspects remain unresolved. Although cristae have been known for almost 60 years and their significance is well appreciated, it is still unclear why IMFM have predominantly tubular cristae, whereas SSM have lamellar cristae; how these arrangements are regulated; and how they are related to mitochondrial function. Also, the precise mechanisms of targeting and assembly of OXPHOS complexes in the cristae membranes remain to be determined.
The increase in mitochondrial permeability leading to cell death and the critical role of MPTP in this process was described over 30 years ago [97]. Several mitochondrial proteins, including VDAC, ANT, CypD, and PiC, have been suggested to be components of MPTP; however, the molecular architecture of MPTP is a matter of open debate. Further rigorous investigations using highly specific modifying agents combined with a genetic approach are required before the molecular composition of MPTP can be conclusively established.
Similarly, multiple mitochondrial channels mediate flux of ions, nucleotides, and proteins across the mitochondrial membranes providing the interface for communication between the site of energy generation and the rest of the cell. Mitochondrial channels are involved in regulation of mitochondrial volume, ATP production, membrane potential and mitochondria-mediated cell death. However, only VDAC, mRyR, and the mitoKV1.3 channel have been identified at the molecular level, whereas the molecular identities of the majority of mitochondrial ion channels remain unknown.
MitoBKCa channel opener NS1619 can inhibit the respiratory activity of isolated cardiac mitochondria [334]. MitoKATP channel opener BMS-180448 has been reported to prevent I/R injury and is currently in clinical trials [335]. Mitochondrial Ca2+ channels also represent attractive targets for the treatment of I/R injury. However, the unknown molecular identities of these channels significantly limit their use as potential targets for cardioprotection. Moreover, the existence of plasma membrane ion channels resembling their mitochondrial counterparts requires searching for selective pharmacological agents applicable for therapeutic targeting of cardiac mitochondria.
Despite the core machineries of mitochondrial fusion and fission are evolutionary conserved from yeast to humans, organization and regulation of mitochondrial dynamics are apparently more complex in mammals than in yeast. This makes the study of the mechanism of mitochondrial dynamics in humans both challenging and potentially very fruitful.
Neurons are particularly sensitive to impairments of mitochondrial dynamics. Defects in MFN2, OPA1, parkin, and PINK1 have been implicated in inherited and age-associated neurodegenerative diseases [336]. Importantly, most of these proteins are even more abundant in cardiomyocytes than in neuronal cells, suggesting their critical roles in cardiac physiology. According to the current consensus, overexpression of the components of fusion machinery leads to interconnected mitochondrial network and plays a cardioprotective role. Overexpression of the fission factors can result in mitochondrial fragmentation contributing to mitochondrial dysfunction and eventually to cell death. Future research has to provide further insight into the molecular mechanism and the role of these processes in the myocardium under normal and pathological conditions.
The mechanisms of mitochondrial transport have also been studied mainly in neurons where this process is particularly important due to the considerable length of neuronal axons. However, based on mitochondria density and high energy demand combined with high expression levels of MIRO1/2 in the myocardium, it is conceivable to assume that similar mechanisms operate in cardimyocytes. Therefore, the key challenge for future studies will be to address the molecular mechanism of mitochondrial trafficking in the heart and its role in cardiac physiology and pathophysiology.
We anticipate that the field will greatly benefit from the mechanistic insights emerging from future studies focused on the molecular structure and dynamics of cardiac mitochondria. Future progress in our understanding of crosstalk between mitochondrial morphology and physiological function is likely to provide novel therapeutic targets and approaches for the more efficient treatment of cardiovascular disorders.
Summary
In mammalian cardiomyocytes, three subpopulations of mitochondria have been described. Intermyofibrillar mitochondria (IMFM) are localized deeper in the cell, longitudinally packed between the sarcomere myofibrils, while subsarcolemmal mitochondria (SSM) are clustered in contact with the sarcolemma. Perinuclear mitochondria (PNM) are typically localized nearby the nucleus and are significantly smaller and have more rounded shape compared to the larger and complex-shaped IMFM.
IMFM and SSM often differ in the structure of their cristae: IMFM have tubular cristae, whereas SSM have lamelliform cristae. Moreover, purified IMFM have almost 50% higher rates of OXPHOS, significantly higher cytochrome c levels and up to threefold higher capacity for Ca2+ uptake than SSM. However, cardiac mitochondria are dynamic organelles, and described differences in morphology of three mitochondrial subpopulations are highly variable depending on physiological and pathological conditions.
The following mitochondrial subcompartments are typically recognized: the mitochondrial outer and inner membranes (MOM and MIM, respectively) with cristae form two aqueous compartments—the space between the MOM and MIM called the intermembrane space (IMS) and the space enclosed by the MIM called the mitochondrial matrix. In addition, the regions of close contacts of the MOM and MIM are called contact sites, while the regions where cristae connect to the MIM are called cristae junctions.
The cristae are not random involutions of the MIM, but they are complex tubular and lamellar structures, which connect to the MIM via narrow tubular regions, cristae junctions. Cristae significantly increase the surface area of the MIM providing sites for assembly of OXPHOS complexes; their numbers are markedly elevated in highly respiratory-active cardiomyocytes.
Mitochondrial membranes contain the major classes of phospholipids characteristic to all cellular membranes, such as phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, phosphatidylserine, phosphatidic acid, and phosphatidylglycerol. In addition to these common membrane phospholipids, mitochondrial membranes contain cardiolipin (CL), which is found predominantly in the MIM.
MOM proteins (account for approximately 4% of the total mitochondrial protein) represent two types of proteins: integral membrane proteins, containing transmembrane domains, and peripheral membrane-associated proteins. They contribute to various essential functions, such as import of fatty acids and proteins into mitochondria, exchange of hydrophilic solutes between the IMS and cytoplasm, and multiple interactions between mitochondria and other organelles. The most abundant protein in the MOM, VDAC (also known as porin) facilitates the transport of ATP/ADP across the MOM.
Unlike the majority of biological membranes, the MIM is characterized by a higher protein content exhibiting a protein to lipid ratio more than 3:1 by weight. Proteins localized in the MIM account for about 21% of the total mitochondrial protein. Since the MIM is highly impermeable to solutes and metabolites, their transport in and out the mitochondrial matrix is mediated by specific membrane-spanning carrier proteins. The MIM also contains three major mitochondrial K+ channels: mitoKATP, mitoKV, and mitoBKCa. Finally, the cristae of highly changeable number and shape contain the electron transport chain complexes I–IV and the F0F1 ATP synthetase (complex V).
Physical contacts between the MOM and MIM, so called contact sites, have been implicated in various mitochondrial processes, such as metabolite and protein transport, bioenergetics, and apoptosis. The presence of the 10–15 nm bridging IMS particles at contact sites suggests that they may represent multiprotein complexes serving various functions. Moreover, contact sites are enriched for CL, which facilitates action of protein complexes involved in transport of small molecules and proteins.
CL represents a relatively small fraction (approximately 16% in rats) of the cardiac phospholipids; however, it is critical for metabolite and protein transport, for OXPHOS, and for the apoptotic cascades. CL is a glycerophospholipid composed of two diacylglycerol phosphate residues and four fatty acid chains. The unique molecular structure of CL facilitates proton-pumping generation of ΔΨm, which is required for ATP synthesis. Alterations in the linoleic acid content and symmetry of CL have been associated with acute myocardial ischemia–reperfusion, heart failure, ageing heart, and diabetes.
CL binds non-covalently to majority, if not all, proteins of the MIM. Among the proteins, which bind CL with high affinity, are the respiratory complexes I, III, IV, and V; the mitochondrial carriers, ANT and PiC; and the IMS-located kinases, mtCK, NDPK-D, and cytochrome c. CL within the MIM facilitates assembly of OXPHOS complexes and higher order supercomplexes. It is also involved in association of ANT with mtCK and NDPK-D facilitating metabolite transport and regulating thereby mitochondrial bioenergetics. CL also interacts with multiple apoptotic proteins, such as Bcl-2 family members, Bid, cytochrome c and caspase-8 contributing thereby to mitochondria-mediated apoptosis. Thus, CL is emerging as a critical integrator, which orchestrates numerous mitochondrial processes ranging from the structural organization of mitochondrial membrane and ATP synthesis to metabolite and protein transport and apoptosis.
The MPTP is a nonspecific channel permeable for most small molecules up to 1.5 kDa, which mediates the increases in the mitochondrial permeability leading to cardiomyocyte death. Its opening induced by Ca2+ overload accompanied by adenine nucleotide depletion, elevated phosphate concentrations, and OS, conditions associated with I/R, results in failure to maintain a barrier to protons. The resulting dissipation of ΔΨm leads to inhibition of ATP synthesis and eventually to bioenergetic failure of the cardiomyocytes. MPTP opening also causes organelle swelling and subsequent rupture of the MOM resulting in the release of mitochondrial apoptogens, such as cytochrome c, Smac/DIBLO, AIF, and endonuclease-G. Ultimately these perturbations result in the apoptotic and/or necrotic death of the cardiomyocytes.
Despite the appreciation of a critical role of the MPTP, the precise molecular structure of the pore remains currently a mystery. Original models have proposed that the MPTP spans both mitochondrial membranes and consists of VDAC in the MOM, ANT in the MIM, and CypD in the matrix. Contact sites appear to be predominant locations of VDAC–ANT–CypD complexes. However, according to the current model, VDAC is no longer a component of the MPTP, the MIM-located PiC, which promotes the Pi reuptake into the matrix, is added to the model as a candidate for the pore-forming subunit, while ANT and CypD represent regulatory subunits of the MPTP.
Mitochondria communicate with the cell via numerous channels. K+, Na+, Ca2+, H+ as well as Pi, nucleotides, and various proteins cross the MOM and MIM in a highly regulated manner to orchestrate a variety of processes ranging from ATP synthesis and mitochondria dynamic organization to cell survival and death.
Ca2+ transport across the mitochondrial membranes mediated by mitochondrial Ca2+ channels plays critical roles in a variety of cellular signaling pathways regulating a myriad of myocardial processes. Several mitochondrial K+ channels, located in the MIM, including mitoKATP, mitoBKCa, mitoKV1.3, and TASK-3, mediate K+ influx into mitochondria, while the K+/H+ exchanger mediates K+ efflux in exchange for protons. Mitochondrial ion channels are involved in regulation of mitochondrial volume, ATP production, ΔΨm, and mitochondria-mediated cell death. Unfortunately, only VDAC, mRyR, and the mitoKV1.3 channel have been identified at molecular level, whereas the molecular identities of the majority of mitochondrial ion channels remain to be a matter of speculation and debate.< div class='tao-gold-member'>Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


