(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Under pathophysiological conditions, reactive oxygen species (ROS) levels may increase and cause cell damage and dysfunction targeting primarily the mitochondria. Using an array of scavenging enzymes and antioxidants, the cardiomyocyte and mainly its mitochondria can neutralize ROS. Importantly, besides its damaging effect on cardiomyocytes, ROS play a central role in a number of signal transduction pathways. Whether the effects of this signaling role are beneficial or harmful may depend upon the location, source, and amount of ROS produced, as well as on the overall redox status of the cell. Indeed, accurate delineation of the downstream signaling pathways involved in ROS accumulation is important in order to improve our understanding of these processes and also to facilitate the development of novel therapies. In this chapter, we will discuss how changes in the cellular redox state may affect cardiac pathophysiology, as well as current and developing therapies.
Introduction
Mitochondria are essential players in the physiology of the cardiomyocyte and play a central role in redox signaling pathways, stress response, myocardial ischemia, heart failure (HF), and hypertrophy, as well as in the aging heart. All cell types including cardiomyocytes are capable of generating reactive oxygen species (ROS), and the major sources of production include mitochondria, the NADPH oxidase (NOX), 5-lipoxygenase (5-LOX) [1], and xanthine oxidases.
Oxygen being the critical terminal electron acceptor in the mitochondrial electron transport chain (ETC), in its absence, the ETC shuts down and the cardiac demands for ATP are not met. Molecular oxygen is also critical in both the formation of nitric oxide (NO), a primary determinant of both vascular tone and cardiac contractility, and the generation of ROS and subsequent induction of oxidative stress (OS), as a by-product of energy metabolism during the sequential acceptance of electrons in the mitochondrial ETC. These short-lived intermediates can act either as important signaling molecules or as inducers of irreversible oxidative damage to proteins, lipids, and nucleic acids; thus, ROS and oxygen exert both beneficial and deleterious effects. ROS have been implicated in the development of agonist-induced cardiac hypertrophy, cardiomyocyte apoptosis, and also in the remodeling processes of the failing heart. Changes in HF phenotype are driven by redox-sensitive gene expression; in this way ROS may act as potent intracellular second messengers. It is important to keep in mind that ROS can be generated in the heart as well as in endothelial tissues by nonmitochondrial enzymes, including the xanthine oxidase (XO), NAD(P)H oxidases, and cytochrome P450 [2]. Moreover, after myocardial infarction (MI), ROS-induced mtDNA damage may result in mitochondrial dysfunction, which will contribute to progressive left ventricular (LV) remodeling and HF. In this chapter, we will discuss how ROS are generated, its critical role in cell damage and signal transduction, its regulation by cellular and mitochondrial antioxidants, and its involvement in cardiac pathophysiology and pathogenesis.
Mitochondrial ROS Production
The major ROS are superoxide radical O2•−, hydrogen peroxide (H2O2), and hydroxyl radical OH•. The superoxide anion (O2•−) is formed when oxygen accepts an electron. Superoxide is in equilibrium with its more reactive protonated form, •HO2, which is favored in acidosis (as occurs with myocardial ischemia). The rates of mitochondrial O2•− generation are known to be inversely correlated with the maximum life-span potential of different mammalian species [3]. The production of the OH•, which is the most reactive form of ROS, is primarily responsible for the damage to cellular macromolecules such as proteins, DNA, and lipids. Formation of the highly reactive OH• results from reactions involving the other ROS species (e.g., the Fenton reaction), in which ubiquitous metal ions, such as Fe (II) or Cu (I), react with H2O2. The high reactivity of the hydroxyl radical and its extremely short physiological half-life of 10−9 s restrict its damaging potential to a small radius from where it is generated since it is too short-lived to diffuse far from its origin [4]. The location of free metal ions can determine the initiation of free radical-caused damage. In contrast, the less reactive superoxide radicals produced in mitochondria can be delivered to the cytosol through anion channels (e.g., VDAC) and thereby may impact sites far from their generation, including activation of transcription factors such as NF-κB among other effects [5]. Similarly, the freely diffusible H2O2 generated in mitochondria can be delivered to the cytosol, where it contributes to increased levels of cellular ROS. It is worth noting that an accurate quantification of mitochondrial ROS flux inside living cells is very difficult because the turnover of several ROS species (particularly the OH• radical) is extremely rapid. Indirect strategies, including the use of gene knockout and overexpression studies [6], have been utilized to demonstrate the presence and involvement of mitochondrial ROS.
Under normal physiological conditions, the primary source of ROS is the mitochondrial ETC, where oxygen can be activated to superoxide radical by a nonenzymatic process. This production of ROS is a by-product of normal metabolism and occurs from electrons produced (or leaked) from the ETC at complexes I, II, and III. Indeed, there is evidence that semiquinones generated within complexes I and III are the most likely donors of electrons to molecular oxygen, providing a constant source of superoxide; [7, 8] also, a supportive role for complex II in ROS production has been suggested [9]. Mitochondrial ROS generation can be amplified in cells with abnormal respiratory chain function as well as under physiological and pathological conditions such as HF, where oxygen consumption is increased.
Besides mitochondria, other cellular sources for generating superoxide radicals include the reactions of oxygen with microsomal cytochrome P450, with reduced flavins (e.g., NOX), usually in the presence of metal ions and also with 5-LOX, one of the enzymes involved in synthesis of leukotrienes from arachidonic acid that in response to stimuli are able to stimulate NOX, mainly growth factors and cytokines. Cytokines lead to membrane ruffling and the generation of superoxide and subsequently to H2O2 through intervention of the small GTPase Rac1 and a superoxide dismutase (SOD) isoform [1]. Moreover, 5-LOX is responsible for IL-4-induced ROS generation and monocyte chemoattractant protein-1 (MCP-1) expression in human vascular endothelial cells (Fig. 10.1). In addition, XO, a primarily cytosolic enzyme involved in purine metabolism, is also a source of superoxide radicals. Particularly, XO activity and its superoxide generation are significantly increased in myocardial post-ischemia/reperfusion damage. In the human myocardium, XO is primarily located in the endothelial cells of capillaries and smaller vessels [10, 11]. Interestingly, the XO inhibitor allopurinol has been found to be protective against the anoxia-related cardiac damage. Recently, Cappola et al. showed a link between XO activity and abnormal cardiac energy metabolism in patients with idiopathic DCM since the inhibition of XO by allopurinol significantly improved myocardial function [12].
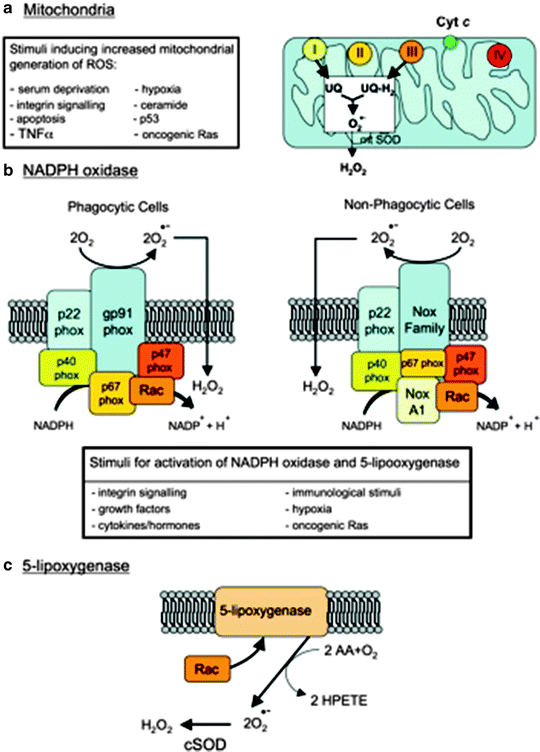
Fig. 10.1
Major cellular sources of reactive oxygen species in living cells. Abbreviations: AA arachidonic acid, Cyt c cytochrome c, cSOD cytosolic SOD, H 2 O 2 hydrogen peroxide, HPETE hydroperoxyeicosatetraenoic acid, mtSOD mitochondrial SOD, NoxA1 NADPH oxidase activator 1, O 2 •− superoxide anion, p53 Tumor suppressor protein 53, phox “phagocyte oxidase” protein, Rac a member of the Rho family of GTPases, Ras small GTPase “rat sarcoma,” ROS reactive oxygen species, SOD superoxide dismutase, TNFα tumor necrosis factor alpha, UQ ubiquinone (adapted from Novo and Parola [1] with permission)
Notably, in the presence of SOD, superoxide radicals can be converted to H2O2 that can further react via the Fenton reaction to form the hydroxyl radical. Superoxide can also react with NO to form peroxynitrite, which is also a highly reactive and hurtful free-radical species (Fig. 10.1).
Effects of ROS on Cardiomyocytes
ROS harmful effects on cardiomyocytes include extensive peroxidative damage to membrane proteins and phospholipids, as well as protein modifications, such as carbonylation, nitration, and formation of lipid peroxidation adducts (e.g., 4-hydroxynonenal [HNE]), which are products of oxidative damage secondary to ROS [13]. Alterations in myocardial respiratory complexes I to V by ROS-mediated nitration, carbonylation, and HNE adduct, with associated decline in their enzymatic activity, have been observed in both in vitro and in vivo studies [14]. Superoxide is also especially damaging to the Fe–S centers of enzymes (e.g., complex I, aconitase, and succinate dehydrogenase). In addition, inactivation of mitochondrial aconitase by superoxide, which generates Fe (II) and H2O2, also increases hydroxyl radical formation through the Fenton reaction [15].
Lipids, and in particular the mitochondrial inner membrane-specific cardiolipin, is a focal target for free-radical damage. A large accumulation of superoxide radicals produced in vitro, with submitochondrial particles from heart, resulted in extensive cardiolipin peroxidation, with a parallel loss of cytochrome c oxidase activity [16, 17]. Oxidative damage also affects nucleic acids and in particular mtDNA by the induction of single- and double-strand breaks, base damage, and modification (including 8-oxoguanosine formation), resulting in the generation of point mutations and deletions [18–20].
In addition, the highly reactive peroxynitrite irreversibly impairs mitochondrial respiration [18] since it inhibits complex I activity, largely by tyrosine nitration of several targeted subunits [20, 21], modifies cytochrome c structure and function [22], affects cytochrome c oxidase activity, inhibits mitochondrial aconitase [23], and causes induction of the mitochondrial permeability transition pore (MPTP) [24]. Some of the effects of peroxynitrite on its mitochondrial targets (e.g., the MPTP) are increased by elevated calcium levels [25]. Notably, the effects of peroxynitrite on mitochondria can be clearly distinguished from the effects of NO, which often are reversible [26]. Therefore, mitochondria, the primary site of intracellular ROS generation, are also the primary locus of its damaging effects. More precisely, damage to mtDNA induces alterations in the mtDNA-encoded polypeptides of the respiratory complexes located in the inner membrane, with consequent decrease of electron transfer and further production of ROS, thus establishing a vicious cycle of OS, mitochondrial dysfunction, and bioenergetic decline.
ROS and Cell Signaling
In addition to the cell-damaging effects of ROS, mitochondrial ROS generation and the OS they engender play a major role in cell regulation and signaling. Oxidative species such as H2O2 and the superoxide anion can be used as potent signals sent from mitochondria to other cellular sites rapidly and reversibly, triggering an array of intracellular signaling cascades leading to diverse physiological end points for the cardiomyocyte, some negative (e.g., apoptosis and necrosis) and others positive (e.g., cardioprotection and cell proliferation). H2O2 produced by mitochondria and sent to the cytosol is involved in several signal transduction pathways, including the activation of c-Jun N-terminal kinase (JNK1) and mitogen-activated protein kinase (MAPK) activities [27, 28] and may affect the regulation of redox-sensitive K+ channels acting on arteriole constriction [29]. The release of H2O2 from mitochondria and its subsequent cellular effects are increased in cardiomyocytes treated with antimycin and high Ca2+ and further enhanced by treatment with coenzyme Q (CoQ). CoQ plays a dual role in mitochondrial generation of intracellular redox signaling by acting both as a prooxidant involved in ROS generation and as an antioxidant by inhibiting the MPTP and cytochrome c release and by increasing ATP synthesis [30]. Increased mitochondrial H2O2 generation and signaling occur with NO modulation of the respiratory chain [25, 28] as well as with the induction of myocardial mitochondrial NO production arising from treatment with enalapril [31]. ROS play a fundamental role in the cardioprotective signaling pathways of ischemic preconditioning, in critical oxygen sensing, and in the induction of stress responses that promote cell survival.
ROS and Cardiac Pathology
Generation of ROS together with impaired antioxidant defenses is observed in myocardial ischemia and reperfusion (I/R) and in the failing heart. During the first moments of reperfusion and myocardial injury, a burst of ROS occurs that is associated with changes in mitochondria (e.g., MPTP opening) [32]. The source of ROS generation during early reperfusion has not been clearly determined; it may be of either mitochondrial or cytoplasmic origin. On the other hand, during ischemia (and likely in the early/acute pathway of ischemic preconditioning), the source of ROS generated clearly involves the mitochondrial ETC, and this may be different than the source of ROS generated in early reperfusion [33, 34]. As it turns out, mitochondria are central in the myocardial failing process since ROS generation by mitochondria impacts the expression of genes encoding key elements of the apoptotic pathway (Fig. 10.2), including MPTP components (i.e., ANT and porin) and the associated mitochondrial creatine kinase in response to HF.
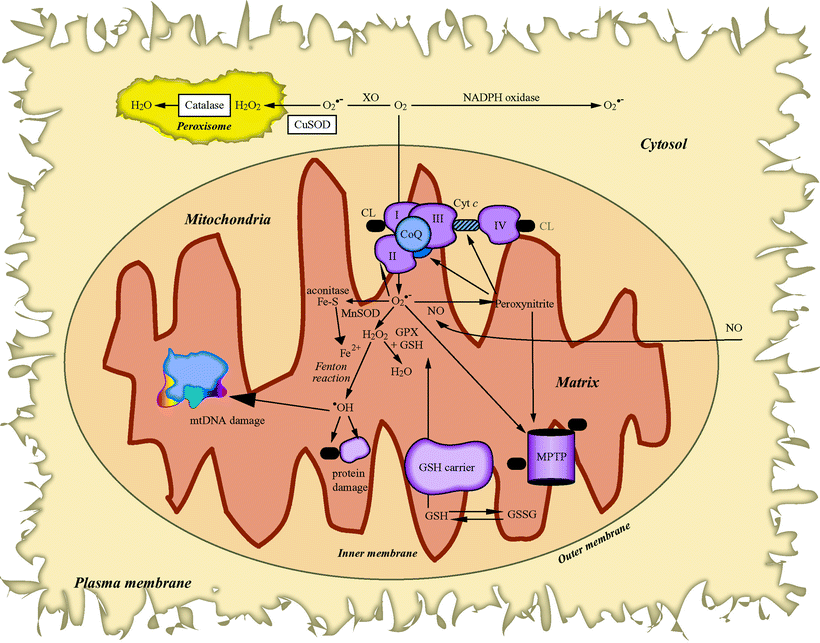
Fig. 10.2
Cellular reactive oxygen species generation and metabolism. Sites of mitochondrial superoxide (O2•−) radical (via respiratory complexes I, II, and III) and cytosolic O2•− generation (by NADPH oxidase or xanthine oxidase, XO) are depicted. Also shown are reactions of the O2•− radical with nitric oxide (NO) to form the highly reactive peroxynitrite, which can target the mitochondrial permeability transition pore (MPTP) opening and the inactivation of mitochondrial aconitase by O2•−. Manganese SOD (MnSOD) (in mitochondria) and copper SOD (CuSOD) (in cytosol) to form hydrogen peroxide (H2O2) are also displayed. The H2O2 is then either further neutralized in the mitochondria by glutathione peroxidase (GPX) and glutathione, in the peroxisome by catalase, or in the presence of Fe2+ via the Fenton reaction, which forms the highly reactive hydroxyl radical (•OH) that can cause severe lipid peroxidation and extensive oxidative damage to proteins and mitochondrial DNA (mtDNA). Other abbreviations: CL cardiolipin, CoQ coenzyme Q, Cyt c cytochrome c, Fe–S iron–sulfur cluster, GSH reduced glutathione, GSSG glutathione disulfide, SOD superoxide dismutase
In HF, cardioprotective interventions may prevent or slow ROS production via antioxidants like MnSOD and target apoptosis. Furthermore, when exposed to an insult, cardiomyocytes are able to mount a cardioprotective response. Also chemical, metabolic, and even physical stressors have been shown to generate cardioprotective responses that share elements of the ischemic preconditioning (IPC) model, and in these responses, mitochondria are the key factors. Besides regulating the cellular bioenergetic supply in the form of adenosine triphosphate (ATP), mitochondria play a multidimensional role that includes participation in the triggering and mediation of cardioprotective responses and the housing and regulation of pivotal early events in the apoptotic pathway. This plasticity enables the organelle to function both as a target and as a player in myocardial signal transduction events and in the generation of ROS in response to a variety of cellular insults providing an appropriate antioxidant response (Table 10.1)
Table 10.1
Mitochondrial functional plasticity allows modulation of the following
Cellular respiration |
Ion (K+, Ca2+) transport |
ATP production |
Cytochrome c release |
Redox state |
ROS generation |
Antioxidant response (e.g., MnSOD, thioredoxin [which functions as electron donor for mitochondrial peroxiredoxin, the ubiquitous enzyme that detoxifies the H2O2 generated by the mitochondrial metabolism], ATP-dependent protease Pim1/LON) |
Cardiac protection with KATP channel openers (e.g., diazoxide and nicorandil) |
Proteome analysis techniques are especially suited to provide a comprehensive picture of the factors regulating protein homeostasis under OS conditions [35–37].
Apart from a direct detoxification of ROS molecules, protein quality control mechanisms are also thought to protect protein functions in the presence of elevated ROS levels. The reactivities of molecular chaperones and proteases remove damaged polypeptides, maintaining enzyme activities, thereby contributing to cellular survival both under normal and stress conditions. Bender et al. [38] have characterized the impact of OS on mitochondrial protein homeostasis by performing a proteomic analysis of isolated yeast mitochondria, determining the changes in protein abundance after ROS treatments. They identified a set of mitochondrial proteins as substrates of ROS-dependent proteolysis. Enzymes containing oxidation-sensitive prosthetic groups like iron/sulfur clusters represented major targets of stress-dependent degradation. Several proteins involved in ROS detoxification were also affected. It was found that ATP-dependent protease Pim1/LON is a major factor in the degradation of ROS-modified soluble polypeptides localized in the matrix compartment. As Pim1/LON expression was induced significantly under ROS treatment, this protease system seems to perform a crucial protective function under OS conditions.
Also, the functional roles of the ATP-dependent protease Pim1/LON and the ClpB-type chaperone Hsp78, both members of the ubiquitous AAA+ (ATPases associated with a wide variety of cellular activities) protein family, have been studied in the context of protein homeostasis processes under normal and stress conditions. It has been found that the coordinated biochemical activities of both Hsp78 and Pim1/LON prevent the accumulation of toxic protein aggregates in mitochondria and thereby indirectly ensure survival of the eukaryotic cell [39].
The time course of MnSOD induction, a mitochondrial-specific protective response to ROS-mediated damage, correlates well with the appearance of ischemic tolerance. However, the activities of other cellular antioxidants, i.e., catalase and CuSOD, are not similarly affected.
In the canine model of pacing-induced HF, there is a marked increase in left ventricular tissue aldehyde levels that suggests elevated free radical-induced damage [40, 41]. The increased levels of OS correlated with the onset of reduced cardiac complex III and V activities. This has been corroborated by measurements of ROS levels in paced compared to unpaced dogs, revealing that both O2•− and OH• radicals generated from mitochondrial-produced H2O2 are increased in the failing heart and correlated with the severity of left ventricular (LV) dysfunction [42, 43]. OS also appears to be involved in the generation of large-scale myocardial mtDNA deletions that have been shown in pacing-induced cardiac failure [40], as well as in studies of ameroid constriction-mediated myocardial ischemia in the dog [44]. Furthermore, neonatal cardiac myocytes treated with TNF-α displayed a significant increase in ROS levels, accompanied by an overall decline in mtDNA copy number and decreased complex III activity [45]. The TNF-α-mediated decline in mtDNA copy number might result from an increase in mtDNA deletions. ROS-induced mtDNA damage, resulting in respiratory complex enzyme dysfunction, contributes to the progression of LV remodeling and failure after MI. In a murine model of MI and remodeling created by ligation of the left anterior descending coronary artery, increased ROS production (e.g., OH• level) was found in association with decreased levels of mtDNA and ETC activities, suggesting impairment of mitochondrial function [46]. In addition, chronic release of ROS has been linked to the development of LV hypertrophy, extracellular matrix (ECM) remodeling, and HF. Interestingly, recent observations have suggested that overexpression of the genes for peroxiredoxin-3 (Prx-3), a mitochondrial antioxidant, or mitochondrial transcription factor A (TFAM), might prevent the decline in mtDNA copy number (likely secondary to mtDNA damage [i.e., increased deletions]) and also the mitochondrial dysfunction observed in HF [47]. Thus, preventing OS and mtDNA damage may be a significant approach in HF treatment.
Besides chronic ROS generation from the mitochondrial organelle, ROS can also derive from nonmitochondrial NADPH oxidase, which in endothelial cells is activated by cytokines, neurohormones, and growth factors (e.g., angiotensin II, norepinephrine, and TNF-α) [48–50]. Furthermore, long-term alterations in cardiac phenotype can be driven by redox-sensitive gene expression, and in this way ROS may act as potent intracellular second messengers. In cardiac myocytes, NADPH oxidase plays a prominent role in the hypertrophic pathway [50–52]. In addition, NADPH oxidase activity is significantly increased in the failing versus nonfailing myocardium [53]. It has been reported that statins (i.e., 3-hydroxyl-3-methylglutaryl coenzyme A [HMG-CoA] reductase inhibitors), by modulating the ROS-generating activity of NADPH oxidase, can inhibit cardiac hypertrophy by cholesterol-independent mechanisms [54, 55]. Statins block the isoprenylation and activation of members of the Rho guanosine triphosphatase (GTPase) family such as Rac1, an essential component of NADPH oxidase. Taken together, it appears that blocking ROS production with statins may be beneficial to patients with myocardial hypertrophy and chronic HF.
Rac1 via NADPH oxidase activation induces myocardial remodeling and dysfunction in diabetic mice. Li et al. [56] reported that Rac1 signaling may be associated with endoplasmic reticulum stress and inflammation, and the targeting inhibition of Rac1 and NADPH oxidase may be a novel therapeutic approach for diabetic cardiomyopathy.
Oxidative Stress in Myocardial Ischemia and HF
Myocardial ischemia can produce changes in the endogenous defense mechanisms against oxygen free radicals, mainly through a reduction in the activity of mitochondrial SOD and a declining tissue content of reduced glutathione [57]. Increased ROS production in mitochondria of circulating white blood cells (WBC) and toxic oxygen metabolite production are exacerbated by readmission of oxygen during postischemic reperfusion. In addition, OS, resulting from both increased ROS generation and diminished antioxidant reserve, promotes the oxidation of thiol groups in proteins and lipid peroxidation leading first to reversible damage and eventually to necrosis.
ROS play an integral role in the genesis and progression of coronary artery disease (CAD) and HF. In the vessel wall, ROS contribute to the formation of oxidized low-density lipoprotein (LDL), a major player in the pathogenesis of atherosclerosis, and ROS-mediated activation of matrix metalloproteinases (MMPs) may play a contributory role in vessel plaque rupture, initiating coronary thrombosis and occlusion [54–56, 58–60].
Increases in hydroxyl and superoxide radicals have been found in animal models of HF. In murine HF created by ligation of the left anterior descending coronary artery for 4 weeks, Ide et al. [46] found that dilatation and decreased LV contractility were accompanied by significant increases in levels of hydroxyl radicals and lipid peroxides. The infarcted LV exhibited diminished activity of respiratory complexes I, III, and IV (enzymes each containing subunits encoded by mtDNA), while the mitochondrial enzymes encoded only by nuclear DNA (e.g., citrate synthase and complex II) were unaffected. Moreover, these investigators using electron spin resonance (ESR) spectroscopy to directly assess ROS levels in the canine model of pacing-induced HF found a significant increase in superoxide anion and hydroxyl radical levels in myocardial submitochondrial fractions [42, 43]. Taken together, these findings suggest that elevated myocardial ROS production was related to functional blockade of the ETC as a result of a marked decrease in mitochondrial respiratory complex activities and also showed a significant positive correlation between myocardial ROS levels and abnormal LV contractility.
Direct measurement of short-lived ROS is extremely difficult, and some observations have shown that increased levels of ROS-mediated damage (e.g., lipid peroxidation, DNA, and protein oxidation) are an indirect index of ROS/OS in several animal models, including pacing-induced HF [2, 40, 61]. In the paced animals, the LV showed increased aldehyde levels and severe reductions in the activity of respiratory complex III and V associated with increased levels of large-scale mtDNA deletions [40].
Both cytosolic antioxidant enzymes (e.g., catalase, SOD1/CuSOD) and mitochondrial-localized antioxidants including SOD2/MnSOD, thioredoxin, and glutathione peroxidase (GPx) can reduce ROS levels. Furthermore, thioredoxin and thioredoxin reductase (TrxR) form an enzymatic antioxidant and redox regulatory system which is implicated in the regeneration of many antioxidant molecules, including ubiquinone, selenium-containing substances, lipoic acid, and ascorbic acid [62].
Some of the most compelling evidence supporting a primary role for mitochondrial ROS and OS in HF are findings with antioxidant genes in transgenic mice. Strains harboring null mutations in either MnSOD or in TrxR2, encoding mitochondrial TrxR, exhibit DCM and HF [63–66]. Li et al. [66] found that mice homozygous for MnSOD deficiency experienced early neonatal death from severe DCM and metabolic acidosis. In addition, these strains displayed severe reduction in myocardial succinate dehydrogenase (complex II) and aconitase (a TCA cycle enzyme) suggesting that MnSOD is required to maintain the integrity of mitochondrial enzymes susceptible to direct inactivation by superoxide.
Mice, in which MnSOD deficiency was targeted to skeletal muscle and heart (i.e., H/M-Sod2 -/- strains), exhibited progressive HF with decreased cardiac contractility by 8 weeks, cardiomegaly by 16 weeks, and death from HF by 22 weeks [66]. This was associated with specific defects in mitochondrial respiration (i.e., severely reduced respiratory complex II and moderately reduced complex I and III activities) and in myocardial mitochondrial ultrastructure. Immunoblot analyses showed increased expression of the SDHA and SDHB subunits from myocardial complex II in H/M-Sod2 -/- mice, with moderate suppression of complex Iα 9, Rieske iron–sulfur protein and Core I subunit of complex III, and α and β subunits of complex V. Mitochondrial superoxide production was also significantly higher in these mice as was mitochondrial (but not cytosolic) lipid peroxidation suggesting that oxidative damage was specifically localized in mitochondria. Furthermore, myocardial ATP production and content were significantly diminished, which may account for the absence of energy-dependent apoptosis in H/M-Sod2 -/- mice. This study showed further evidence that ROS and OS are intimately linked to the progression of HF since the administration at 8 weeks of age of the antioxidant MnSOD mimetic (MnTBAP) improved cardiac contractility and ameliorated the overall phenotype.
Overexpression of the antioxidant GPx in transgenic mice inhibited the development of LV remodeling and failure after MI and was associated with the attenuation of myocyte hypertrophy, apoptosis, and interstitial fibrosis [67]. Moreover, overexpression of catalase (primarily a peroxisomal-localized enzyme) targeted to the mitochondria increased overall mouse longevity, diminished OS- and ROS-mediated mitochondrial protein and mtDNA damage, and delayed the onset of aging-mediated cardiac pathology including subendocardial interstitial fibrosis, vacuolization of cytoplasm, variable myofiber size, hypercellularity, collapse of sarcomeres, mineralization, and arteriosclerosis. Interestingly, it mattered in which subcellular compartment the overexpressed catalase was localized [68]. While there was little evidence of benefits from nuclear or peroxisomal-localized catalase, the benefits observed on mitochondrial-localized catalase activity was striking.
Clinically, a clear link between OS/ROS and chronic ventricular dysfunction has only been established in anthracycline-mediated cardiomyopathy and in cases with alcoholic cardiomyopathy. On the other hand, it remains unclear whether ROS or OS has a pathophysiologic role in the majority of patients with congestive HF or cardiomyopathy due to ischemic, hypertensive, valvular, or idiopathic causes [69]. Superoxide anions as assessed by electron paramagnetic resonance spectroscopy (EPR) with an O2•− spin trap were reported to increase more than twofold in the failing ventricular myocardium from patients undergoing transplant [70]. Moreover, despite increased MnSOD mRNA levels, marked decline in mitochondrial-localized MnSOD protein and activity was present. Both increased ROS levels and decreased antioxidant response would be expected to lead to enhanced OS in the failing heart, which in turn may result in increased transcription of antioxidant enzymes. That excessive ROS/OS in HF may serve as a potent trigger for changes in specific nuclear gene expression and may also underlie the programmatic shift in mitochondrial bioenergetic function previously discussed and its role as a catalyst in myocyte remodeling.
In vitro and in vivo animal studies have shown that in the failing heart, ROS may affect several components of the cardiac phenotype and its remodeling, including contractile function, interstitial fibrosis, endothelial dysfunction, and myocyte hypertrophy. ROS contribution to the remodeling processes is shown in a number of ways: (1) activation of MMPs, which participate in the reconfiguration of the ECM; (2) acting as signaling molecules in the development of compensatory hypertrophy; and (3) as contributors to myocyte loss via apoptosis signaling.
Excessive production of NO is also implicated in the pathogenesis of chronic HF [71, 72]. Uncoupling of constitutive nitric oxide synthase (NOS) leads to overproduction of superoxide (O2•−) and peroxynitrite (ONOO−), two extremely potent oxidants. Peroxynitrite, produced from the reaction of highly reactive NO with the superoxide anion, impairs cardiovascular function through multiple mechanisms, including activation of MMPs and nuclear enzyme poly (ADP-ribose) polymerase (PARP). Increased OS resulting from the overproduction of superoxide also mediates the dysregulation of S-nitrosylation of proteins at specific cysteine residues by reactive nitrogen species (RNS), a more selective modification of proteins that is found with protein oxidation. This redox mechanism has been demonstrated to lead to abnormal myocardial excitation–contractility and vascular reactivity [73, 74].
ROS in the Aging Heart
The effects of ROS on mitochondria may be responsible for alterations of mitochondrial function observed during various abnormal states associated with OS and aging.
Increased OS in the aging and failing heart is mainly related to increase electron leak through impaired complex I, II, and III (Fig. 10.3), although both normal aging human and animal hearts have increased levels of ROS and reactive nitrogen species (RNS). The balance between ROS-/RNS-generating enzymes and scavenger enzyme systems can be disrupted in pathological conditions, including degenerative diseases [75], diabetes mellitus [76], endothelial dysfunction, atherosclerosis, and hypertension [77].
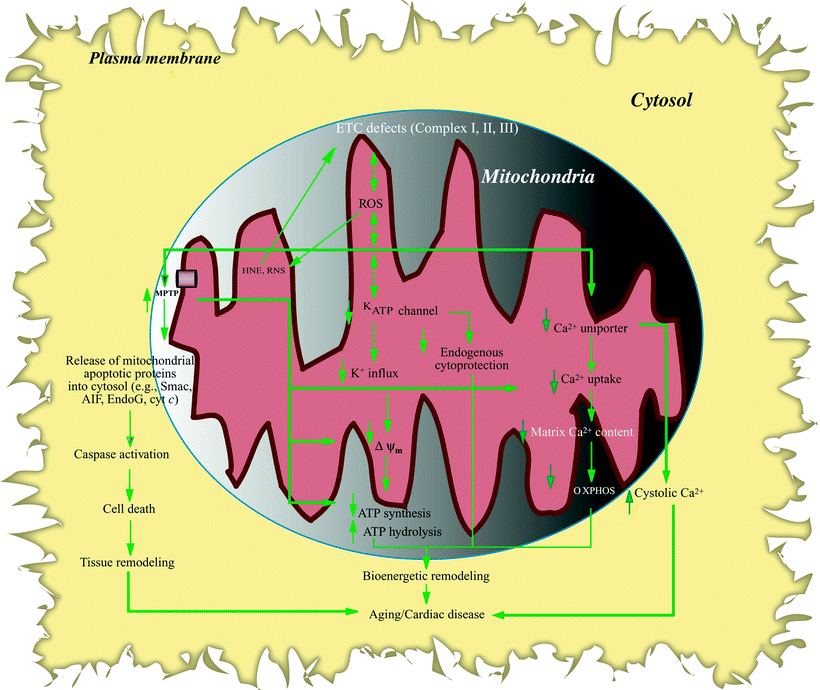
Fig. 10.3
Flow chart of remodeling resulting from aging/heart failure modulation of mitochondrial ion function. Mitochondrial electron transport chain (ETC) defects result in electron leak, reactive oxygen species (ROS) overproduction followed by formation of reactive nitrogen species (RNS), and lipid peroxidation (e.g., 4-hydroxynonenal, HNE). Increased oxidative stress, in turn, damages mitochondrial ion channel function. Sensitized mitochondrial permeability transition pore (MPTP) causes loss of mitochondrial Ca2+, decreased Ca2+ uptake, and reduced mitochondrial membrane potential (ΔΨm) which in turn reduces oxidative phosphorylation (OXPHOS) and ATP synthesis; increased MPTP also promotes increased mitochondrial vulnerability to Ca2+ loading and release of apoptogenic factors (e.g., apoptosis-inducing factor (AIF), cytochrome c (cyt c)) from mitochondria which sets into motion the apoptotic pathway including caspase activation, leading to cell death. Mitochondrial ATP-regulated K+ channel (KATP) is also targeted in aging and heart failure (HF), resulting in reduced cardiac tolerance to stress and loss of preconditioning cytoprotection; moreover, decreased K+ influx and electrochemical gradient results in diminished OXPHOS. Reduced Ca2+ uniporter activity not only decreases mitochondrial Ca2+ uptake, dampening Ca2+ stimulatory effects on bioenergetic metabolism, but also exaggerates intracellular Ca2+ disturbance, leading to elevated cytosolic Ca2+ levels. KATP and MPTP channel modulation can trigger ROS production, which can further target ETC activities, as does increased HNE (indicated by double-headed arrows). Other abbreviations: EndoG endonuclease-G, Smac Smac/DIABLO proapoptotic protein. Solid vertical arrows indicate reduced or increased levels, whereas unfilled arrows denote relationship within a pathway
Antioxidant mechanisms may be enhanced in the myocardium with aging; however, these compensatory mechanisms appear not to be completely effective in stemming ROS accumulation. Increased protein oxidation, lipid oxidation, and mtDNA damage increase with aging, whereas chronic ROS stress acts as a trigger of matrix fibrosis. With aging and in the failing aging heart, increases in oxidative damage to proteins by ROS or RNS are characterized by protein carboxylation and nitration.
Mitochondria are not only considered the major source of ROS, but they are also the targets of ROS during the aging process, and increased mitochondrial ROS formation is expected to cause mitochondrial protein oxidative damage in the senescent animals [77–79].
ANT is one of the mitochondrial proteins most susceptible to ROS during aging [80], and ANT oxidative damage is mostly displayed as carbonylation. Increased ANT carbonyl modification has also been found in aged houseflies [80] as well as in aging mammalian animals [81–83]. ANT contains three redox-sensitive cysteine residues on loops projecting into the matrix compartment. These residues are particularly susceptible to oxidation by sulfenic acid moieties and mixed disulfides if the normally high intramitochondrial glutathione (GSH)/glutathione disulfide (GSSG) ratio is not maintained during periods of OS [84]. Indeed, GSH/GSSG ratio is significantly reduced during aging [85–89].
Importantly, ANT carbonyl modification is associated with a reduced capability to exchange AD/ATP across the inner membrane, which may cause the inhibition or uncoupling of OXPHOS and the collapse of ΔΨm, turning mitochondrial ATP synthesis into hydrolysis (Fig. 10.4). ANT oxidation may also shift from its native state as a gated pore (mediating ADP/ATP exchange) into a nonselective pore, allowing free permeation of small ions and metabolites across the mitochondrial inner membrane. Furthermore, in the mitochondrial matrix, ANT conformation conversion is affected by cyclophilin-D (CyP-D). CyP-D contains a relatively high proportion of polyunsaturated fatty acids that could be oxidized by ROS to generate highly reactive lipid fragments such as 4-hydroxy-2-nonenal (HNE) and 4-hydroxyhexenal (HHE). These lipid peroxidation by-products may modify specific thiol groups on ANT leading to adduct formation, thereby inhibiting ANT activity [90], and also increasing CyP-D binding to ANT [91, 92] thereby destabilizing ANT and sensitizing MPTP to calcium [93].
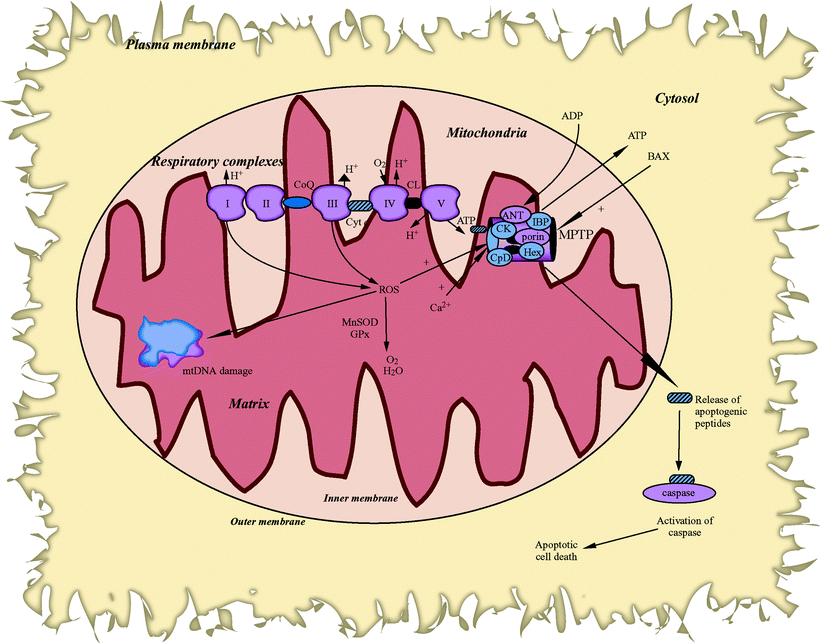
Fig. 10.4
Generation of reactive oxygen species by mitochondria produces mitochondrial DNA damage and impacts the expression of genes encoding key elements of the apoptotic pathway. Abbreviations: ANT adenine nucleotide translocator, BAX Bcl-2-associated X protein, CK creatine kinase, CL cardiolipin, CoQ coenzyme Q, CpD cyclophilin D, Cyt c cytochrome c, GPx glutathione peroxidase, Hex hexokinase, IBP isoquinoline binding protein, MnSOD manganese superoxide dismutase, MPTP mitochondrial permeability transition pore, mtDNA mitochondrial DNA, ROS reactive oxygen species
With aging, VDAC is also particularly vulnerable to oxidative damage because the tyrosine residue is subject to conversion into 3-nitrotyrosine (3-NT) by RNS including NO and ONOO−, which are normally produced under physiologic conditions and increased with aging. Nitration of tyrosine can compromise the functional and/or structural integrity of the target protein [94]. Interestingly, proteomic analysis using 2D gel electrophoresis combined with nanoelectrospray ionization tandem mass spectrometry (NSI-MS/MS) technique showed an age-dependent nitration of VDAC in mitochondria [95]. Increased VDAC nitration is very likely related to excessive OS in mitochondria since similar changes have been found in diabetic hearts which exhibit increased OS [96]. In permeabilized HepG2 cells [97], it is not known if oxidative modification of tyrosine residues directly alters the property of VDAC or sensitizes VDAC to its regulators; however, it has been found that O2•− could induce VDAC-dependent rapid and massive cytochrome c release and enhance MPTP sensitivity to Ca2+ in permeabilized HepG2 cells [97].
Ca2+ is the key trigger for MPTP formation, possibly by binding to the negatively charged cardiolipin head groups on the inner face of the inner membrane, thereby disrupting the stabilizing interactions between cardiolipin and ANT [98]. Since mitochondrial Ca2+ content tends to decrease with aging, the impaired mitochondrial Ca2+ handling and reduced threshold of MPTP to Ca2+ may be more critical than Ca2+ itself in mediating MPTP in the aging failing heart. For instance, in response to stress such as ischemia/reperfusion, heart mitochondrial Ca2+ significantly increased (about twofold greater with senescence). Taking these findings together with the evidence of increased lipid peroxidation (HNE formation) in senescent hearts on reperfusion, it seems that the aging hearts are increasingly vulnerable to reperfusion damage brought about by increased mitochondrial Ca2+ [99], increased creatine kinase nitration [100, 101], age-dependent decreased hexokinase [99], and decreased nucleotide pool [81], which may also facilitate the formation of ANT-VDAC complex, sensitize MPTP, or induce the permeability transition in aging and in the aging failing heart.
It is worth noting that the mitochondrial proteolytic system plays an important role in preserving the integrity of the organelle in the presence of OS. An important factor for protein maintenance is enzymatic reversal of oxidative modifications and/or protein degradation, and failure of these protein maintenance systems may be a critical component of the aging process. The ATP-stimulated mitochondrial Lon protease is a highly conserved protease found in prokaryotes and the mitochondrial compartment of eukaryotes and is believed to play an important role in the degradation of oxidized mitochondrial matrix proteins [102]. Age-dependent decreases in the activity and regulation of this proteolytic system may result in accumulation of oxidatively modified and dysfunctional proteins and loss in mitochondrial viability.
Oxidative Stress and Apoptosis
Both the generation of ROS and the onset of apoptotic cell death are important and often connected events in cell homeostasis. Increased OS and ROS accumulation can lead to myocyte hypertrophy, interstitial fibrosis (through matrix metalloproteinases activation), and apoptosis. Apoptosis can be blocked or delayed by treatment with antioxidants and thiol reductants [103]. Moreover, overexpression of antioxidant proteins (e.g., MnSOD, GPx, and metallothionein) can block apoptotic progression [67, 104, 105]. Attenuation of apoptosis by overexpression of the antiapoptotic protein Bcl-2 was associated with protection against ROS and OS [106, 107]. Cells from transgenic mice containing ablated genes encoding antioxidant proteins (e.g., GPx) have both increased OS levels and increased apoptosis [106, 108].
Agents that induce apoptosis (e.g., TNF-α) also promote high levels of mitochondrial-generated ROS [109]. In addition, ROS production can target an array of signal transducers (e.g., JNK, TNF-α, and MAPK), which interface with the apoptotic machinery. This includes both activation of the proapoptotic proteins from Bcl-2 family and apoptosis mediators (e.g., TNF-α), as well as the modulation of their gene expression by activation of specific transcription factors (e.g., NF-κB).
It is worth noting that an important model system has emerged to study the induction of OS and apoptosis using H2O2-treated cultured cardiomyocytes [110]. This approach allowed molecular and biochemical appraisal of cellular and mitochondrial events presaging, accompanying, and following the induction of cardiomyocyte apoptosis. In addition, it allowed the study of short-lived signaling intermediates, as well as the use of various treatments (e.g., antioxidants) to stem the development of cardiomyocyte OS and apoptotic progression [111, 112]. Not all cardiomyocyte apoptosis is triggered by OS and ROS generation. Gathered observations have shown no evidence of ROS (or NO) involvement in the palmitate-mediated induction of apoptosis in neonatal rat cardiomyocytes [113], illustrating the complex, multiforked, and parallel nature of signals in the apoptotic pathway.
Mitochondrial Nitric Oxide
Oxygen serves as the critical terminal electron acceptor in the ETC; in its absence, the ETC shuts down, and cardiac demands for ATP are not met. Molecular oxygen is also central in both the formation of NO, a primary determinant of both vascular tone and cardiac contractility, and in the generation of ROS (and subsequent OS) as a significant by-product of energy metabolism during its sequential acceptance of electrons in the mitochondrial ETC. The end results of NO/redox disequilibrium have implications for cardiac and vascular homeostasis and may result in the development of atherosclerosis, myocardial tissue remodeling, and hypertrophy. ROS/RNS generation is also attributed to the transit from hypertrophic to apoptotic phenotypes, a possible mechanism of myocardial failure [114].
Endothelial regulation of vascular tone is mediated mainly by NO, and abnormal production and/or distribution of ROS and RNS in blood creates oxidative and/or nitrosative stresses in the failing myocardium and endothelium. As previously noted, the uncoupling of constitutive NOS leads to increased generation of superoxide (O2•−) and peroxynitrite (ONOO−). Peroxynitrite affects cardiovascular function and will contribute to the pathogenesis of cardiac and endothelial dysfunction associated with MI, chronic HF, diabetes, atherosclerosis, hypertension, aging, and various forms of shock. Moreover, pharmacological inhibition of XO-derived superoxide formation, as well as neutralization of peroxynitrite, or inhibition of PARP provides significant benefit in various forms of cardiovascular injury [72]. Increased OS resulting from the overproduction of superoxide mediates the dysregulation of proteins via S-nitrosylation at specific cysteine residues by RNS, a more selective modification of proteins than found with protein oxidation. This redox mechanism has been found to lead to altered myocardial excitation–contractility and vascular reactivity [73, 74].
Antioxidant Defenses
In the heart, a large number of vital pathways, including energy metabolism, survival and stress responses, apoptosis, inflammatory responses, and oxygen sensing, are regulated by ROS and cellular redox states. There is extensive evidence showing that the role of ROS in ischemia/reperfusion (I/R) injury and the role that antioxidants therapy may play vary significantly depending on whether we use whole organ and animal models or isolated cell models; findings are often contradictory in providing insights into why antioxidant clinical trials have frequently shown mixed results.
The powerful cell-damaging ROS can be neutralized by an array of protective antioxidant scavenger enzymes, as well as by various lipid- and water-soluble compounds including ascorbic acid, glutathione, thioredoxin, and α-tocopherol. Antioxidant enzymes are located in a variety of cellular compartments including the mitochondria (e.g., MnSOD, GPx, TrxR), peroxisomes (e.g., catalase), microsomes (e.g., cytochrome P450), and cytosol (e.g., CuSOD). It is generally accepted that levels of antioxidants in myocardial mitochondria are significantly lower than in liver mitochondria, although the majority of opinion is that the antioxidant capacity of the heart is sufficient to handle normal levels of ROS production but insufficient to meet the greater accumulation of ROS that occurs in myocardial ischemia [115, 116].
In rats, a mitochondrial isoform of catalase with low specific activity has been found [116, 117]. This mitochondrial catalase activity was detected in the heart but not in liver or skeletal muscle and appears to increase during caloric-restricted (CR) diets and in the diabetic heart [118–120]. The role that this enzyme plays has not been fully determined, although it probably participates in the prevention of excessive lipid peroxidation in myocardial ischemia [121]. On the other hand, mitochondria-specific catalase has not been found in the heart of transgenic mice even after overexpression of the catalase gene [122].
While superoxide dismutases by the formation of H2O2 catalyze the removal of superoxide radicals, GPx catalyzes the breakdown of H2O2 to water and GSSG by using GSH as shown in Fig. 10.1. Since GPx is located in both the mitochondria and cytosol, H2O2 can be removed from either compartment depending on the availability of glutathione. A small fraction of the total cellular pool of GSH is sequestered in mitochondria by the action of a carrier that transports GSH from cytosol to the mitochondrial matrix [123]. Upon exposure to increased exogenous ROS, isolated, perfused rat hearts are rapidly depleted of their antioxidant reserves, including those of SOD and GSH, rendering them more vulnerable to the action of oxidative injury [124].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


