(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Mitochondria are both sources and targets of reactive oxygen species, and there is increasing evidence that mitochondrial dysfunction may be a significant mechanism by which cardiovascular risk factors lead to the formation of vascular lesions. Recent studies have focused on the role that mitochondria could play in atherogenesis since mitochondria are both important sources and targets of ROS. This is further suggested by the mitochondrial dysfunction theory which postulates that excess release of ROS and RNS from mitochondria can contribute to the inflammatory vascular reaction, leading to the development of atherosclerotic lesions. Also, common cardiovascular risk factors encountered in daily clinical practice could be involved in this process by adversely affecting the function of endothelial mitochondria, and mitochondrial dysfunction may be the most important unifying mechanism explaining the atherogenic action of major cardiovascular risk factors. The molecular mechanisms by which atherosclerotic risk factors could lead to mitochondrial dysfunction and subsequent vascular impairment will be reviewed in this chapter.
Introduction
Atherosclerosis is the key underlying mechanism for cardiovascular disease and its clinical consequences constitute the leading causes of morbidity and mortality in the Western world. There is wide acceptance that atherosclerosis is an inflammatory disease associated with lipid and protein oxidation in the vascular wall [1, 2]. Increased level of low-density lipoprotein (LDL) is associated with increased risk of coronary artery disease [3]. Oxidative modification of LDL, and its transport into the subendothelial space of the arterial wall at the sites of endothelial damage, is considered an initiating event for atherosclerosis [4, 5]. Oxidative modification of LDL results from the interaction of reactive oxygen species (ROS) and reactive nitrogen species (RNS), produced from vascular wall cells and macrophages [4] with LDL. The resulting increased oxidative and nitrosooxidative stress induces endothelial dysfunction by impairing the bioactivity of endothelial nitric oxide and promotes leukocyte adhesion, inflammation, thrombosis, and smooth muscle cell proliferation—processes that are known to exacerbate atherosclerosis.
Recent studies have focused on the role that mitochondria could play in atherogenesis. In fact, mitochondria are both important sources and targets of ROS [6]. The mitochondrial dysfunction theory postulates that excess release of ROS and RNS from mitochondria can contribute to the inflammatory vascular reaction, leading to the development of atherosclerotic lesions [7, 8]. In fact, increased mitochondrial ROS production causes endothelial dysfunction, vascular smooth muscle cell (VSMC) proliferation, and apoptosis of VSMCs and macrophages, with ensuing atherosclerosis (ATS) lesion progression and possible plaque rupture [9].
Common cardiovascular risk factors encountered in daily clinical practice could be involved in this process by adversely affecting the function of endothelial mitochondria, and growing evidence supports the hypothesis that mitochondrial dysfunction may be the most important unifying mechanism explaining the atherogenic action of major cardiovascular risk factors [2, 7, 9]. This chapter will therefore review the molecular mechanisms by which atherosclerotic risk factors could lead to mitochondrial dysfunction and subsequent vascular impairment.
Mitochondrial Dysfunction in Atherosclerosis
Mitochondria can serve as both the sources as well as the targets of ROS. There is increasing evidence to support the concept that mitochondrial dysfunction is a highly relevant intermediate mechanism by which cardiovascular risk factors lead to atherosclerosis. Mitochondrial function is required for normal vascular cell growth and function. Mitochondrial dysfunction can result in apoptosis, leading to plaque rupture and acute coronary syndromes. Much of what is known on ROS generation and modulation comes from studies in cultured cells and animal models.
Oxidative Dysfunction
Oxidative damage to the mitochondria is caused primarily by the ROS generated by mitochondria themselves [10, 11], principally due to the release of electrons by the coenzymes NADH and FADH2 into the electron transport chain. Mitochondria possess at least nine known sites that are capable of generating superoxide anion, a progenitor ROS. Mitochondria also possess numerous ROS defense systems that are much less studied [12]. In addition, a significant amount of ROS can be produced by the enzymes alpha-ketoglutarate dehydrogenase and monoamine oxidase located in the mitochondria outer membrane (MOM) [12, 13].
The deleterious effects resulting from the formation of ROS in mitochondria are neutralized to a certain extent by various antioxidant systems. Under normal conditions there is a balance between ROS formation and degradation. However, when the antioxidant defenses become insufficient and cannot convert ROS to H2O2 rapidly enough, oxidative damage accumulates in the mitochondria [14]. On the other hand, free fatty acids (FFAs) can decrease the mitochondrial generation of ROS under conditions of reverse electron transport, due to their uncoupling action. Under conditions of forward electron transport, FFAs can actually increase ROS production [15]. This dual effect was investigated in rat heart and liver mitochondria under conditions of forward and reverse electron transport. Under conditions of the forward electron transport, i.e., with pyruvate plus malate and with succinate (plus rotenone) as respiratory substrates, polyunsaturated fatty acid, arachidonic, and branched-chain saturated fatty acid, phytanic, increased ROS production in parallel with a partial inhibition of the electron transport in the respiratory chain, most likely at the level of complexes I and III. A linear correlation between stimulation of ROS production and inhibition of complex III was found in rat heart mitochondria. This effect on ROS production was further increased in glutathione-depleted mitochondria. Under conditions of the reverse electron transport, i.e., with succinate (without rotenone), unsaturated fatty acids, arachidonic and oleic, straight-chain saturated palmitic acid, and branched-chain saturated phytanic acid strongly inhibited ROS production. This inhibition was partly abolished by the blocker of ATP/ADP transfer, carboxyatractyloside, thus indicating that this effect was related to uncoupling (protonophoric) action of fatty acids. Therefore, it has been concluded that in isolated rat heart and liver mitochondria, functioning in the forward electron transport (ET) mode, unsaturated fatty acids (FAs) and phytanic acid increase ROS generation by inhibiting the ET transport and by changing membrane fluidity. Only under conditions of reverse ET, FAs decrease ROS generation due to their uncoupling action [15].
Although relatively controversial [16], nitric oxide (NO) production does seem to occur in mitochondria through different pathways [17, 18]. In isolated mouse brain, heart, and liver, mitochondria produced reactive nitrogen species (RNS), which was inhibited by catalysts of peroxynitrite decomposition but not by NO inhibitors. Disrupting the organelles or withdrawing respiratory substrates markedly reduced ROS production. Inhibition of complex I abolished the diaminofluorescein (DAF) signal, restored by complex II substrates [18]. Inhibition of the respiratory complexes downstream from the ubiquinone/ubiquinol cycle or dissipating the proton gradient had no effect on DAF fluorescence [18]. The NO produced in the mitochondria [17] may counteract superoxide at multiple levels. For example, it can rapidly scavenge superoxide via direct radical–radical reaction to form peroxynitrite, a potent oxidant [19–22], capable of decreasing the activity of complex I by forming S-nitrosothiols [23]. This in turn reduces the generation of mitochondrial ROS. Furthermore, NO indirectly facilitates the superoxide scavenging by stabilizing cytochrome c and preventing its leakage from mitochondria [24].
Asymmetrical dimethyl l-arginine (ADMA), an endogenous NO synthase inhibitor [25], can also lead to increased mitochondrial ROS levels [26]. In both recombinant human endothelial NO synthase (eNOS) and pulmonary arterial endothelial cells (PAEC), ADMA increased NO synthase (NOS) uncoupling. This eNOS uncoupling increased 3-nitrotyrosine levels preferentially in the mitochondria of PAEC due to a redistribution of eNOS from the plasma membrane to the mitochondria. The increase in nitration in the mitochondria was found to induce mitochondrial dysfunction as determined by increased mitochondrial-derived reactive oxygen species and decreased generation of ATP. Finally, the decrease in ATP resulted in a reduction in the chaperone activity of HSP90, resulting in a decrease in its interaction with eNOS. Thus, increased levels of ADMA cause mitochondrial dysfunction and a loss of heat shock protein-90 chaperone activity secondary to an uncoupling of eNOS [26]. Mitochondrial ROS production can also be increased by the mitochondrial p66Shc protein, which favors cytochrome c release, dissipation of mitochondrial transmembrane potential, and apoptosis [27, 28]. p66(Shc−/−) and WT mice were utilized to investigate the effects of high-fat diet on both systemic and tissue oxidative stress and the development of early vascular lesions. Computer-assisted image analysis revealed that chronic 20% high-fat treatment increased the aortic cumulative early lesion area by 20% in WT mice and only by 3% in p66(Shc−/−) mice. Early lesions from p66(Shc−/−) mice had less content of macrophage-derived foam cells and apoptotic vascular cells compared to WT. In p66(Shc−/−) mice, but not WT mice, there was a significant reduction of systemic and tissue oxidative stress, as assessed by isoprostanes, plasma low-density lipoprotein oxidizability, and the formation of arterial oxidation-specific epitopes [28].
Mitochondrial dysfunction is also implicated in the increased susceptibility to ischemic injury, in part, because of opening of the mitochondrial permeability transition pore (MPTP) [29–31]. The molecular components of MPTP include adenine nucleotide translocator (ANT) in the inner mitochondrial membrane, voltage-dependent anion channel in the outer mitochondrial membrane, cyclophilin D in the matrix, as well as regulatory molecules such as benzodiazepine receptor, hexokinase, and creatine kinase [32]. Transient MPTP opening causes depolarization of mitochondrial membrane potential, whereas longer opening leads to matrix swelling and outer mitochondrial membrane rupture. The latter causes the release of proapoptotic molecules within the intermembrane space into cytosol, leading to cell death via caspase-dependent and caspase-independent mechanisms [33]. It has been hypothesized that ischemia/reperfusion promotes mitochondrial permeability transition pore (MPTP) in two phases: (1) MPTP priming during ischemia occurs as progressive inner mitochondrial membrane leak is accompanied by depressed electron transport in the setting of fatty acid accumulation and loss of cytochrome c and antioxidants, and (2) triggering of MPTP at reperfusion is determined by the interplay of mitochondrial membrane potential (ΔΨm) with mitochondrial matrix Ca2+, ROS, and pH [33]. Consistent with this concept, mitochondrial depolarization has been implicated in hyperglycemia-induced apoptosis of human aortic endothelial cells (Fig. 14.1) [34]. Decrease in ANT activity associated with ischemia and inhibition of both ANT activity and oxidative phosphorylation evident during reperfusion may contribute to the development of heart failure [35]. Overexpression of mitochondrial superoxide dismutase 2 (SOD2, MnSOD) offered protection against ischemia/reperfusion injury [36], whereas heterozygous deficiency of this enzyme impaired postischemic recovery of the myocardium in mice [37]. All together, these data support the role of mitochondrial function in protection against ischemia/reperfusion injury.
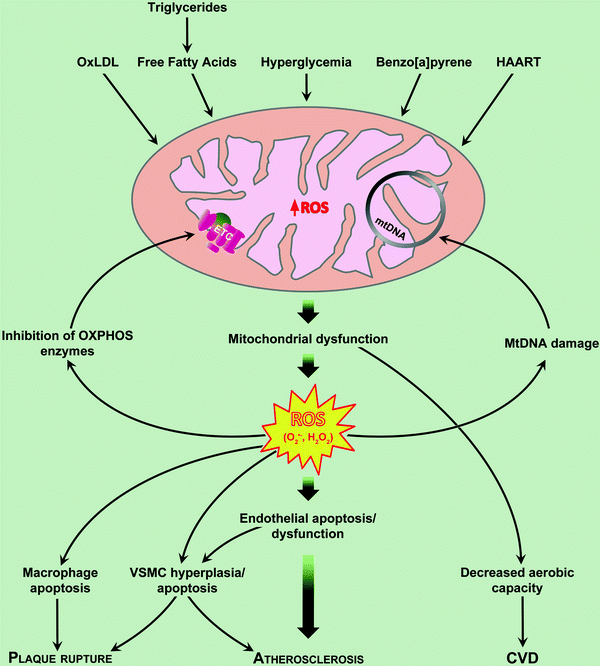
Fig. 14.1
Mechanisms of mitochondrial dysfunction in atherosclerosis. Mitochondria-derived reactive oxygen species (ROS) in the endothelial cell act as physiologic intracellular messengers and as cause of oxidative damage. Oxidized LDL (oxLDL) induces apoptosis of all cells involved in atherogenesis. HAART Highly Active Anti-Retroviral Therapy CVD. Cardiovascular disease. See text for further details
Oxidative Damage
Mitochondrial DNA (mtDNA) is likely the most sensitive cellular target of ROS as it is located close to the inner mitochondrial membrane where ROS are produced. Moreover, mtDNA is small in size and is not protected by histone proteins as is the case for nuclear DNA [38]. Many different types of oxidative DNA lesions have been described, ranging from base adduct modifications to single- and double-strand breaks [39]. The hydroxyl radical can remove protons from deoxyribose, thus producing a sugar radical and inducing strand breaks and release of the affected DNA base [40]. Moreover, the hydroxyl radical can also abstract a proton from the methyl group of thymine and add it to the C4, C5, and C8 positions of purines, generating hydroxyl adduct radicals [41].
The contribution of mitochondrial oxidant generation and DNA damage to the progression of atherosclerotic lesions has been examined in human arterial specimens and atherosclerosis-prone mice [7]. Mitochondrial DNA damage not only correlated with the extent of atherosclerosis in human specimens and aortas from apolipoprotein E(−/−) mice but also preceded atherogenesis in young apolipoprotein E(−/−) mice. Apolipoprotein E(−/−) mice deficient in manganese superoxide dismutase, a mitochondrial antioxidant enzyme, exhibited early increases in mitochondrial DNA damage and a phenotype of accelerated atherogenesis at arterial branch points. Thus, mtDNA damage correlates with the extent of atherosclerosis, suggesting also that mitochondrial dysfunction may promote atherogenesis [42]. Indeed, many of the DNA modifications can contribute to aging, cancer, and neurodegenerative diseases [43], as well as to other pathophysiological conditions [44]. Damage to mtDNA can have a greater impact on cellular function than damage to nuclear DNA [45].
The accumulation of mtDNA mutations can cause cellular dysfunction by altering oxidative phosphorylation and Ca2+ homeostasis, inducing further oxidative stress and a defective turnover of mitochondrial proteins, and affecting the susceptibility to apoptosis [14]. In particular, the mtDNA-encoded respiratory enzymes can increase electron leak and ROS production, with subsequent enhanced oxidative stress and further damage to mitochondria [46]. A vicious cycle may therefore be generated, leading to progressive accumulation of ROS and oxidative damage to mtDNA [42].
Increased production of ROS in mitochondria can damage lipids, proteins, and mtDNA. Among these, as alluded to earlier, mtDNA is likely the most sensitive to physiologically relevant ROS-mediated damage. Preferential increase in mtDNA damage (compared with transcriptionally inactive nuclear β-globin gene), decrease in steady-state levels of mtDNA-encoded mRNA transcripts, mitochondrial protein synthesis, membrane potential, and total cellular ATP pools were observed in vascular smooth muscle cells (VSMCs) and endothelial cells exposed to ROS in cell cultures [19]. 4-Hydroxynonenal, an end product of membrane lipid peroxidation implicated in the pathogenesis of atherosclerosis, induces VSMC apoptosis through mitochondrial dysfunction and increased production of ROS [47]. By contrast, increased ROS production attributable to haploinsufficiency of SOD2 isoform reduced aconitase activity in both basal and agonist-stimulated conditions and increased VSMC proliferation [48].
Mitochondrial Dysfunction in Conditions Associated with Atherosclerosis
These include diabetes mellitus and dyslipidemia; other risk factors such as hypertension and aging will be discussed in other chapters.
Diabetes Mellitus
Type 2 diabetes mellitus is an increasingly prevalent risk factor for atherosclerosis and vascular disease [49]. ROS participates in the onset and development of diabetes. ROS can induce inactivation of the signaling pathway between the insulin receptor and the glucose transporter system, leading to insulin resistance [50]. On the other hand, in addition to being a possible effect of ROS production, diabetes is also a source of oxidative stress, with resulting atherogenic effect [51]. Hyperglycemia induces superoxide generation in endothelial cells, and most of this superoxide may be produced by mitochondria [52]. Electron transfer and oxidative phosphorylation are uncoupled, resulting in superoxide formation and inefficient ATP synthesis.
A key mechanism in atherogenesis is endothelial dysfunction, characterized by decreased NO bioavailability and the development of an inflammatory phenotype that promotes atherosclerosis [53]. Endothelial dysfunction contributes to the development and progression of atherosclerosis in patients with diabetes. There is a growing knowledge of the importance of altered mitochondrial dynamics in diabetes [54]. Mitochondria undergo cycles of fusion to form networks and fission to form smaller individual mitochondria [54, 55]. Proteins controlling fusion include mitofusin (Mfn)-1, Mfn2, and optic atropy-1 (Opa1). Fission is regulated by dynamin-related protein-1 (Drp1) and fission-1 (Fis1). Fusion may be beneficial by allowing the distribution of metabolites, proteins, and DNA throughout the network. At the end of their life cycle, dysfunctional mitochondria and damaged mitochondrial components are eliminated by fission and subsequent autophagy [56]. Under pathological conditions, including diabetes mellitus, fission is increased and autophagy is impaired, leading to a loss of mitochondrial networks, accumulation of small dysfunctional mitochondria, and increased mitochondrial ROS [57]. Recent studies have demonstrated a loss of mitochondrial networks under hyperglycemic conditions in a variety of cell types, including islet cells [58], hepatocytes [57], skeletal muscle cells [59], circulating blood mononuclear cells [60], and endothelial cells [61].
A recent report has investigated the contribution of altered mitochondrial dynamics to endothelial dysfunction in diabetes mellitus [62]. In venous endothelial cells isolated from subjects with diabetes, mitochondrial fragmentation as well as increased expression of fission-1 protein was observed compared healthy control subjects. In cultured human aortic endothelial cells exposed to glucose, a similar loss of mitochondrial networks and increased expression of Fis1 and Drp1, proteins required for mitochondrial fission, was observed. Altered mitochondrial dynamics was associated with increased mitochondrial ROS production and a marked impairment of agonist-stimulated activation of eNOS and cGMP production. Silencing Fis1 or Drp1 expression with siRNA blunted high glucose-induced alterations in mitochondrial networks, ROS production, eNOS activation, and cGMP production. An intracellular ROS scavenger provided no additional benefit, suggesting that increased mitochondrial fission may impair endothelial function via increased reactive oxygen species. These novel findings implicate increased mitochondrial fission as an important contributing mechanism for endothelial dysfunction in diabetes.
Dyslipidemia
Apoptotic endothelial cells, vascular smooth muscle cells, T lymphocytes, and macrophages are present in atherosclerotic lesions [63, 64], and their number increases with the progression of lesions, suggesting that apoptosis plays a critical role in plaque erosion and rupture [65, 66]. Oxidized LDL (oxLDL) induces apoptosis of all cells involved in atherogenesis [67, 68] (Fig. 14.1), and mitochondrial-dependent pathways play a critical role in this process. OxLDL-induced apoptosis of human umbilical vein endothelial cells (HUVECs) is mediated by dysfunction of mitochondrial membrane potential and the release of cytochrome c into cytosol, and suppression of apoptosis by cyclosporin A, an antiatherogenic agent, correlated with the prevention of mitochondrial dysfunction [69]. More recently, it was shown that oxLDL-induced vascular cell apoptosis involves two distinct calcium-dependent mitochondrial pathways [70]. The first is mediated by activation of the cysteine protease calpain; release of tBid, a truncated form of the proapoptotic Bcl-2 family member Bid; opening of the MPTP; release of cytochrome c; as well as the subsequent activation of caspase-3. The second pathway is mediated by the release of apoptosis-inducing factor, which is cyclosporine insensitive and caspase independent (Fig. 14.2). It has also been demonstrated that mitochondrial-derived O2•− is essential for the oxidation of LDL in vitro [71]. When human endothelial cells (ECs) were incubated with the mitochondrial respiratory chain inhibitors—antimycin A or rotenone or with the uncoupler (carbonyl cyanide m-chlorophenylhydrazone)—rhodamine 123 uptake and subcellular distribution were altered, and concomitantly superoxide anion production and LDL oxidation were strongly decreased. These observations suggest that mitochondrial function is required, directly or indirectly, for the production of superoxide anion and the subsequent LDL oxidation by human vascular ECs.
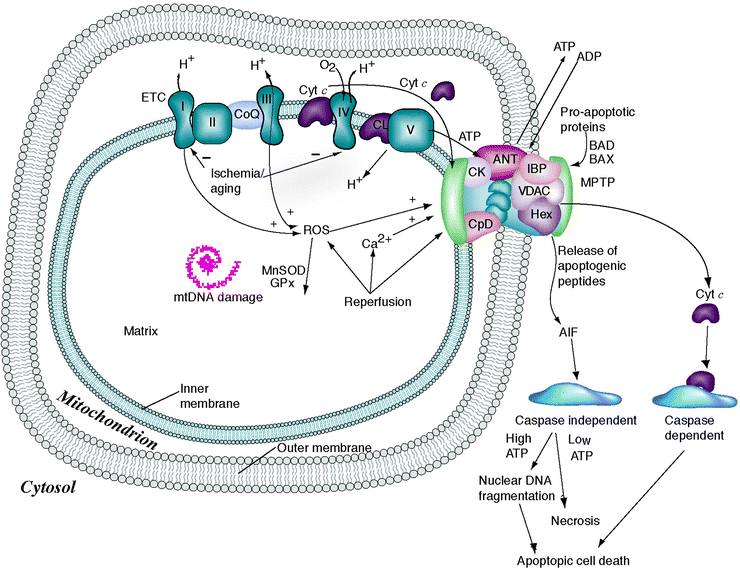
Fig. 14.2
Mitochondrial permeability transition pore and apoptosis. Oxidized LDL (OxLDL)-induced vascular cell apoptosis involves two distinct calcium-dependent mitochondrial pathways. The first is mediated by activation of the cysteine protease calpain; release of tBid, a truncated form of the proapoptotic Bcl-2 family member Bid; opening of the MPTP; release of cytochrome c; as well as the subsequent activation of caspase-3. The second pathway is mediated by the release of apoptosis-inducing factor, which is cyclosporine insensitive and caspase independent. See text for details
Macrophages in advanced atherosclerotic lesions accumulate excess free cholesterol, which is a potent inducer of apoptotic death [72]. Free cholesterol loading of mouse peritoneal macrophages induced apoptosis by decreasing mitochondrial membrane potential, inducing cytochrome c release, activating caspase-9, and increasing the levels of the proapoptotic protein Bax [73]. OxLDL also induced lysis of human macrophages by promoting mitochondrial dysfunction, and scavengers of peroxide radicals restored mitochondrial membrane potential and prevented macrophage lysis [74]. In addition, increased oxidative stress in mitochondria is evident from the induction of transcription and expression of SOD2 in human macrophages incubated with oxLDL [75]. Consistent with this in vitro observation, SOD2 activity and concentration of reduced form of glutathione were higher in atherosclerotic intima compared with the media of the aorta of heritable hyperlipidemic rabbits, but a significant inverse correlation of these two with lesion size was also observed. TUNEL-positive nuclei were present in the macrophages of these atherosclerotic aortas, and exposure to oxLDL induced apoptosis in human macrophages. Hypercholesterolemia significantly increased mtDNA damage and protein nitration of heart homogenates, indicating that atherosclerotic risk factors induce mitochondrial damage and dysfunction [76]. MtDNA copy number in leukocytes is redox sensitive [77] and low in hyperlipidemic patients [78]. Taken together, these data suggest that dyslipidemia-induced mitochondrial damage and dysfunction not only induce atherosclerotic lesion formation but also affect lesion composition and progression.
Conclusions
Oxidative stress, which may be induced by commonly known cardiovascular risk factors, plays an important role in the development of atherosclerosis. Mitochondria are both sources and targets of reactive oxygen species, and there is growing evidence that mitochondrial dysfunction may be a relevant intermediate mechanism by which cardiovascular risk factors lead to the formation of vascular lesions. Mitochondrial DNA is probably the most sensitive cellular target of reactive oxygen species. Damage to mitochondrial DNA has been correlated with the extent of atherosclerosis. Several known cardiovascular risk factors are demonstrated causes of mitochondrial damage. Oxidized low-density lipoprotein and hyperglycemia can induce the production of reactive oxygen species in mitochondria of macrophages and endothelial cells. On the other hand, reactive oxygen species may favor the development of type 2 diabetes mellitus, through the induction of insulin resistance.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


