CHAPTER 43 The Posterior Mediastinum
ANATOMY
The posterior mediastinum, also called the paravertebral compartment, is a defined space between the posterior pericardium and the anterior spinal ligament including the paravertebral gutters. According to the traditional four-compartment model, the mediastinum is divided into anterior, middle, posterior, and superior compartments. The posterior mediastinum is bounded superiorly by the inferior border of the T4 vertebral body.1,2
In the traditional three-compartment model without a superior division, the entire length of the spine is included in the posterior mediastinum.2 The diaphragm is the common inferior border for all three compartments. Within these boundaries, the posterior mediastinum contains the esophagus, descending aorta, sympathetic chain and vagus nerves, thoracic duct, azygos and hemiazygos veins, fat, and lymph nodes (Fig. 43-1).1
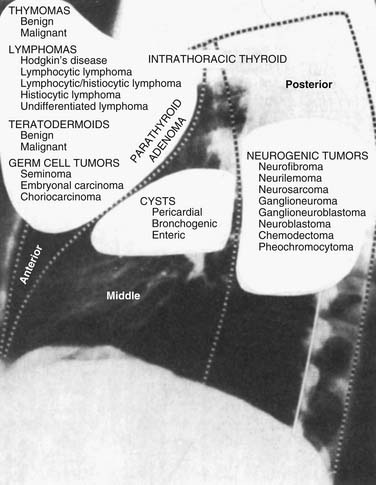
EPIDEMIOLOGY
Great differences exist between children and adults with respect to the location of mediastinal masses.3 In adults, 65% of the lesions arise in the anterosuperior, 10% in the middle, and 25% in the posterior compartments; this distribution is reversed in children, in whom 25% of lesions arise in the anterosuperior, 10% in the middle, and 65% in the posterior compartments. In general, the incidence of posterior mediastinal lesions is higher in children, whereas anterior lesions predominate in adults.
DIAGNOSIS
Computed tomography (CT) is the imaging modality of choice for the posterior mediastinum because of its ability to localize masses and to determine somewhat the extent of invasion. CT can often distinguish among the various masses and identify their origin, allowing correct diagnosis based on radiographic imaging.4 Although magnetic resonance imaging (MRI) has been cited as useful in particular circumstances, such as with dumbbell tumors, cysts, and extramedullary hematopoiesis, CT remains the superior imaging modality for most posterior mediastinal masses.5 Mediastinal sonography, although less expensive and comparable to CT in the diagnosis of many mediastinal masses, is not useful in the posterior mediastinum.6
The decision to sample a mediastinal mass is not straightforward. Biopsy before resection is not necessary in some cases and potentially harmful in others. The likelihood of a positive finding on biopsy depends on the presence of local symptoms and the location and extent of the lesion. Lesions in the posterior mediastinum require either fine-needle aspiration (CT guided) or a thoracoscopic approach.7,8 If these procedures are not possible because of location in the paravertebral gutters, a limited posterolateral thoracotomy should be used to obtain adequate tissue for diagnosis.9,10
SURGICAL APPROACHES TO THE POSTERIOR MEDIASTINUM
Multiple standard approaches have been described to reach the posterior mediastinum. These include cervical, paravertebral, thoracotomy, transabdominal, and thoracoscopic (Fig. 43-2).
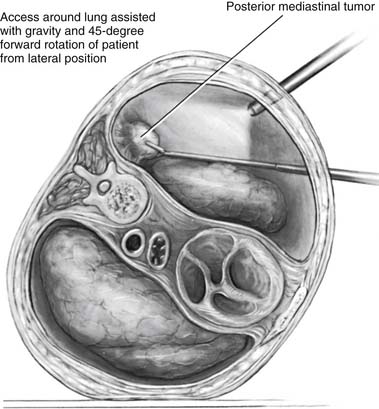
Figure 43–2 Thoracoscopic resection of a posterior mediastinal tumor.
(Reprinted with permission from Davis RD Jr, Sabiston DC Jr. Textbook of surgery. Philadelphia: WB Saunders; 1997. p. 1915.)
The posterior mediastinum can also be accessed transabdominally through the esophageal hiatus in various antireflux procedures, in hiatal hernia repairs, and during the transhiatal esophagectomy. A combined cervical approach with transabdominal mobilization allows the surgeon to remove the esophagus extrapleurally without traversing the pleural cavities or bony thorax.11
Currently, the decision to perform thoracoscopic surgery for posterior mediastinal lesions remains dependent on the surgeon’s experience and comfort with video-assisted procedures; but for biopsy, this should be the procedure of choice.8,12,13 Zierold and Halow14 conducted a retrospective review that described use of thoracoscopic approaches for posterior mediastinal neurogenic masses. A total of 29 patients (13 men, 16 women), aged 26 to 68 years, who underwent a thoracoscopic resection were identified. Preoperative imaging included chest radiography and CT in all patients and MRI in 15 of 29 patients (52%). All tumors were located in the posterior mediastinum without preoperative evidence of invasion or malignancy. Conversion to an open procedure was required in 12 of 29 (41%) patients (mini-thoracotomy in 11, posterolateral thoracotomy in 1). Tumor size necessitating conversion to an open procedure (mean = 4.79 cm) and tumor size amenable to thoracoscopy alone (mean = 3.84 cm) were not significantly different (P < .09). They concluded that thoracoscopic resection could be performed successfully, regardless of tumor type or size; however, malignant lesions, local invasion, and tumors larger than 5 cm may require an open procedure. Cardillo and associates15 recently detailed their experience with 93 patients who underwent resection of a posterior mediastinal neurogenic tumor. The tumor was resected with a videothoracoscopic approach in 57 patients; 44 underwent video-assisted thoracoscopic surgery only, and 13 required conversion to an open approach. Mean operative time was significantly shorter in the video-assisted thoracoscopic surgery group, as was the median postoperative hospital stay and postoperative pain. At a mean follow-up of 73 months, no recurrences were noted.
LESIONS OF THE MEDIASTINUM
Neurogenic Tumors
Neurogenic tumors of the thorax commonly occur in the posterior mediastinum and primarily affect young adults and children.16 In recent decades, although these tumors continue to be the most common malignant neoplasm in children, they have become less common than tumors of the anterior mediastinum (thymomas or lymphomas) in adults. They now represent approximately 15% of all mediastinal masses in adults. Furthermore, in adults, the malignancy rate of neurogenic tumors is less than 10%. In children, fully 50% of these lesions are malignant.16 Takeda and colleagues17 reviewed their experience with 146 patients with intrathoracic neurogenic tumors treated during a 50-year period. In this series, there were 60 pediatric patients and 86 adults. There were 51 ganglioneuromas, 37 schwannomas, 30 neurofibromas, 18 neuroblastomas, 5 ganglioblastomas, and 5 others. Of these lesions, 136 were located in the posterior mediastinum, but only 13 patients (8.9%) had intraspinal extension of tumor; 84% of adults and 60% of children were asymptomatic at the time of presentation and 20.5% of the lesions were malignant, these occurring predominantly in the first 5 years of life.
Neurogenic tumors originate from embryonic neural crest cells around the spinal ganglia and from either sympathetic or parasympathetic components (Box 43-1). The differential diagnosis for neurogenic tumors arising from the intercostal nerves includes neurofibroma, neurilemoma, and neurogenic sarcoma. Sympathetic ganglia tumors include ganglioneuroma, ganglioneuroblastoma, and neuroblastoma. Pheochromocytomas can occur from paraganglia cells. Neurogenic tumors rarely arise from the phrenic and vagus nerves.
Neurogenic tumors that arise from intercostal nerves are typically neurilemomas or neurofibromas.18 Neurilemomas (schwannomas) are the most common neurogenic tumors.19,20 On histologic examination, they appear as well-encapsulated, firm, gray-tan masses.21 They tend to occur in one of two morphologic patterns: either an organized architecture with a cellular palisading pattern of growth (Antoni’s type A) or a loose reticular pattern (Antoni’s type B).22 Neurofibromas are poorly encapsulated, with a random arrangement of spindle-shaped cells.23 Associated with neurofibromatosis type 1, these tumors tend to form in the paravertebral gutters (Fig. 43-3). Both neurilemomas and neurofibromas can occur as manifestations of von Recklinghausen’s disease and can degenerate into neurosarcoma if they are left untreated.
Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma are tumors of the sympathetic nervous system that arise from primitive sympathetic ganglia and are referred to collectively as neuroblastic tumors.24 They arise wherever sympathetic tissue exists and may be seen in the neck, posterior mediastinum, adrenal gland, retroperitoneum, and pelvis. The three tumors differ in their degree of cellular and extracellular maturation; immature tumors tend to be aggressive and occur in younger patients (median age, just under 2 years), whereas mature tumors occur in older children (median age, approximately 7 years) and tend to behave in a benign fashion.24
Ganglioneuromas usually manifest as a benign tumor composed of gangliocytes and mature stroma. They usually appear at an early age located in the paravertebral region and are the most common neurogenic tumors occurring in childhood. Ganglioneuroblastomas are composed of both mature gangliocytes and immature neuroblasts and have intermediate malignant potential.25
Neuroblastomas are also seen in children; more than 75% of the cases occur in children younger than 4 years. On histologic examination, these tumors are composed of small, round immature cells organized in a rosette pattern. They are highly invasive, and by the time diagnosis is made, they have often metastasized to the regional lymph nodes, bone, brain, liver, and lung. Interestingly, however, they may, in certain patients, have a relatively benign course, even when metastatic. Symptoms at time of diagnosis typically include cough, dysphagia, chest pain, and occasionally paraplegia. At times, they may manifest with paraneoplastic syndromes such as profuse watery diarrhea secondary to vasoactive intestinal protein syndrome, opsoclonus-polymyoclonus syndrome, and pheochromocytoma-like syndrome. Features such as DNA content, tumor proto-oncogenes, and catecholamine synthesis influence prognosis, and their presence or absence aids in categorizing patients with neuroblastoma as high, intermediate, or low risk. Treatment consists of resection and, usually, chemotherapy. Despite recent advances in treatment, including bone marrow transplantation, neuroblastoma in most patients remains a relatively lethal tumor, accounting for 10% of pediatric cancers but 15% of cancer deaths in children.24
A CT scan can help elucidate the tumor type and extent of tumor involvement (Figs. 43-4 and 43-5).5,24 Neurogenic tumors from peripheral nerves appear as well-defined round or oval masses that are noncalcified in the paravertebral gutter. Neurilemomas have variable enhancement with either homogeneity or heterogeneity.26 With enhanced CT, these tumors demonstrate variable attenuation, depending on their histology. Neurofibromas are usually homogeneous, low-attenuation lesions on unenhanced CT. Enhanced CT demonstrates homogeneous enhancement or early central blush. Malignant nerve sheath tumors show variable attenuation.22
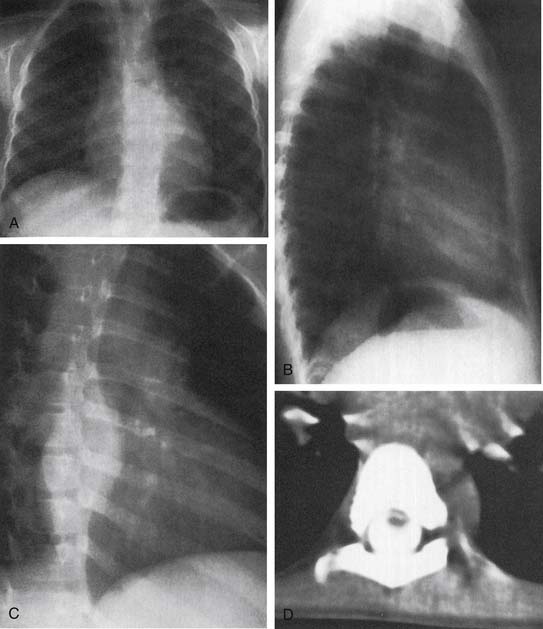
Figure 43–4 A to C, Chest films of a ganglioneuroblastoma. D, CT image of tumor extension into the spinal column.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


