CHAPTER 58 The Pharmacologic Management of Heart Failure
PATHOPHYSIOLOGY OF HEART FAILURE
The term heart failure does not refer to a single entity; rather, it denotes a syndrome that is characterized by signs or symptoms of intravascular volume overload or manifestations of inadequate tissue perfusion. It is the end result of a variety of cardiac injuries and the ensuing pathologic remodeling that impair the heart’s ability to fill with or to eject blood; it may originate from a wide range of disorders of the myocardium, pericardium, endocardium, or intracardiac valves (Table 58-1). Heart failure can be categorized pathophysiologically in several ways.
Table 58–1 Common Causes of Heart Failure
| Myocardial |
| Ischemia or infarction |
| Viral myocarditis |
| Idiopathic cardiomyopathy |
| Hypertrophic cardiomyopathy |
| Hypertension |
| Toxins (alcohol, cocaine, chemotherapeutic agents) |
| Infiltrative diseases (amyloidosis, hemochromatosis) |
| Infectious (Lyme disease, Chagas’ disease) |
| Peripartum cardiomyopathy |
| Thyroid dysfunction |
| Metabolic abnormalities (thiamine or selenium deficiency) |
| Valvular |
| Aortic stenosis |
| Aortic regurgitation |
| Mitral stenosis |
| Mitral regurgitation |
| Arrhythmic |
| Tachycardia-mediated cardiomyopathy |
| Pericardial |
| Constrictive pericarditis |
Heart failure may be classified symptomatically on the basis of its clinical severity (Table 58-2). Despite the varied causes and classifications of heart failure, the manifestations all reflect intravascular volume overload, inadequate tissue perfusion, or their combination. A thorough explanation of all these forms of heart failure is beyond the scope of this chapter. The following discussion, therefore, focuses primarily on left ventricular systolic failure in both the acute and chronic setting.
Table 58–2 Clinical Classifications of Heart Failure
| New York Heart Association Classification | |
| Class I | Symptoms only with greater than usual activity |
| Class II | Asymptomatic at rest but with symptoms during normal activities |
| Class III | Asymptomatic at rest but with symptoms during minimal exertion |
| Class IV | Symptoms at rest |
| American College of Cardiology/American Heart Association Classification | |
| Stage A | Patients with structurally normal hearts, asymptomatic, but at risk for the development of heart failure due to the presence of risk factors (e.g., hypertension, coronary artery disease, diabetes) |
| Stage B | Patients with structurally abnormal hearts (e.g., left ventricular systolic dysfunction, left ventricular hypertrophy, valvular dysfunction, prior myocardial infarction) but without symptoms of heart failure |
| Stage C | Patients with structurally abnormal hearts and current or prior symptoms of heart failure |
| Stage D | Patients with end-stage heart failure symptoms not responsive to standard therapy |
Acute Heart Failure
The response of the cardiovascular system to the onset of myocardial dysfunction and the pathophysiologic mechanisms underlying the subsequent progression to heart failure depends in large part on the acuity of the dysfunction. An acute cardiac insult results in a series of hemodynamic alterations that account for the clinical manifestations of left ventricular failure. This occurs irrespective of whether the initial insult depresses myocardial contractility (systolic dysfunction) or impairs ventricular filling (diastolic dysfunction). The cascade begins with a rise in left ventricular end-diastolic pressure. This elevated pressure is transmitted to the left atrium and subsequently to the pulmonary venous and capillary system. The increased intravascular pressure results in transudation of fluid into the pulmonary interstitium, where it interferes with gas exchange, resulting in hypoxemia and dyspnea. In addition, there is often an associated reduction in cardiac output resulting in inadequate delivery of blood to the arterial system with resultant organ hypoperfusion.
The heart’s response to these hemodynamic alterations is the activation of several compensatory mechanisms (Table 58-3). A rapid, generalized activation of the adrenergic system occurs and is associated with a withdrawal of parasympathetic tone. Direct sympathetic stimulation of the heart and β-adrenergically induced release of epinephrine and norepinephrine from the adrenal glands result in tachycardia and an increase in myocardial contractility, both of which serve to augment cardiac output. Catecholamine-induced peripheral arterial vasoconstriction redirects the available cardiac output away from relatively nonessential organs (i.e., skin, skeletal muscle, gut, kidney) and helps maintain sufficient blood pressure to ensure adequate perfusion of more vital organs (i.e., heart and brain). Furthermore, β-adrenergic stimulation of the juxtaglomerular apparatus in the kidneys results in the release of renin and activation of the renin-angiotensin system. The angiotensin II thus produced is a potent vasoconstrictor and acts in concert with the direct α-adrenergic stimulation of the vasculature to maintain blood pressure. The reduced renal blood flow from depressed cardiac output and redistribution of blood volume results in activation of renal baroreceptors. This further stimulates renin release and augments sympathetic activation, thereby contributing to vasoconstriction.
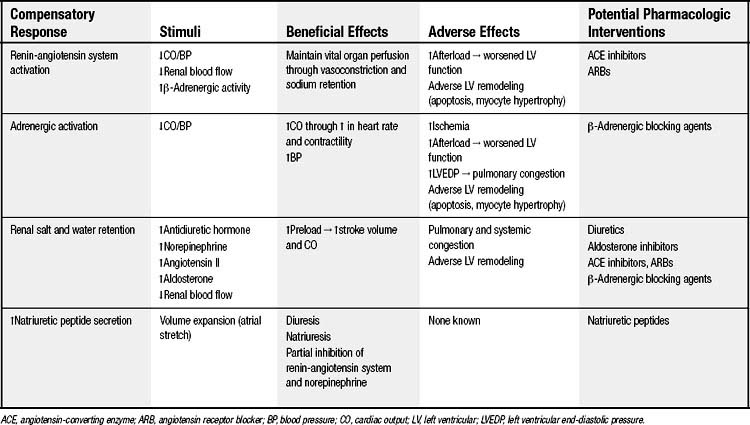
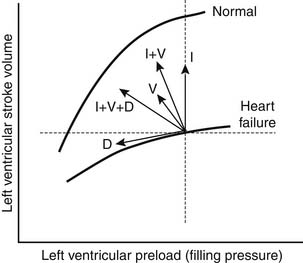
In addition to producing these hemodynamic alterations, acute heart failure is characterized by marked sodium and water retention. This occurs through a variety of mechanisms. Angiotensin II directly promotes the reabsorption of sodium in the proximal nephron and indirectly promotes the reabsorption of sodium from the distal nephron, this latter effect being mediated through angiotensin II–induced release of aldosterone from the adrenal cortex. Furthermore, angiotensin II and norepinephrine stimulate hypothalamic release of arginine vasopressin, resulting in further vasoconstriction and free water reabsorption. These changes produce an expansion of intravascular volume and augmentation of venous return, thereby increasing ventricular end-diastolic volume (preload). The increased preload results in an increase in stroke volume by the Frank-Starling mechanism (Fig. 58-1) and thereby helps support the cardiac output.
Chronic Heart Failure
In combination, the aforementioned mechanisms serve a compensatory role in acute heart failure, helping to maintain cardiac output and blood pressure to allow adequate perfusion of vital organs. These compensatory mechanisms may initially be adequate to allow clinical stability, and the patient may subsequently go through a stage of asymptomatic ventricular dysfunction, maintained in part by chronic stimulation of the adrenergic and renin-angiotensin systems. As heart failure progresses, the hemodynamic overload induces changes in the shape and size of the ventricle, a process known as ventricular remodeling. The specific changes that occur depend in part on the hemodynamic stressors facing the ventricle. In predominantly pressure-overloaded conditions (e.g., hypertension, aortic stenosis), there is a rise in systolic wall stress that results in left ventricular hypertrophy. If this hypertrophy is insufficient to normalize the wall stress, dilation occurs. Under conditions of volume overload (e.g., aortic or mitral insufficiency), there is a rise in diastolic wall stress that induces ventricular dilation. This dilation in turn results in increased systolic wall stress (by the Laplace relationship) and subsequent hypertrophy. These hypertrophic changes help maintain systolic wall stress within a normal range and help preserve ventricular contractile function. However, with continued hemodynamic overload, there is progressive ventricular dilation, eventuating in the development of a dilated, spherical heart. This altered ventricular morphology produces less efficient ventricular contraction, may induce mitral regurgitation due to annular dilation and malcoaptation of the valve leaflets, and is associated with an adverse prognosis.
The stimuli that induce ventricular remodeling are varied and the mechanisms underlying the remodeling process are complex (Table 58-4). It appears that increased wall stress (resulting from ventricular dilation and increased afterload) and neurohormones (i.e., β-adrenergic and renin-angiotensin systems), vasoactive peptides (e.g., endothelin), and cytokines (e.g., tumor necrosis factor α) mediate remodeling. These factors may have direct effects on the cardiac myocytes or may act indirectly through stimulation of second-messenger systems and thereby induce a variety of changes in myocyte structure and function. On a cellular level, myocyte hypertrophy results from the replication of sarcomeres either in parallel (producing ventricular hypertrophy) or in series (producing ventricular dilation). Alterations in the expression of various contractile proteins occurs with reexpression of fetal genes and reduced expression of adult contractile genes, resulting in abnormalities of calcium handling and excitation-contraction coupling. Chronic stimulation of the sympathetic system is accompanied by a reduction in the density of β-adrenergic receptors in the myocardium and an uncoupling of the receptors from their intracellular mediators. This results in a blunted response of the failing myocardium to either endogenous (e.g., exercise) or exogenous (e.g., dopamine or dobutamine) adrenergic stimulation.
Table 58–4 Factors Involved in Ventricular Remodeling
| Stimulants of Ventricular Remodeling | Molecular and Cellular Events That Mediate Ventricular Remodeling |
|---|---|
As the remodeling process develops, the neurohormonal effects of the β-adrenergic and renin-angiotensin systems on the peripheral vasculature and renal salt and water handling continue. The intense vasoconstriction, while maintaining flow to vital organs, contributes to hypoperfusion of the kidneys and progressive renal dysfunction. The augmented preload and cardiac output resulting from sodium and water retention help maintain circulating blood volume and tissue perfusion. However, the associated increase in end-diastolic pressure and increased ventricular wall stress contribute to progressive ventricular remodeling and result in pulmonary and systemic venous hypertension and precipitation of congestive symptoms. Thus, these initially compensatory changes become deleterious in the chronic setting. The inhibition of these processes offers not only a mechanism for the treatment of heart failure but also the potential to reverse the adverse remodeling seen in the chronic state (see Table 58-3).
PHARMACOLOGIC AGENTS USED IN THE TREATMENT OF HEART FAILURE
The majority of pharmacologic agents used for the treatment of heart failure effect benefit either directly by interfering with the hemodynamic alterations described before or through inhibition of the neurohormonal activation underlying these alterations (see Fig. 58-1). These medications fall into several categories: diuretics, vasodilators, inotropic agents, and neurohormonal inhibitors. Diuretics act to reduce preload (leftward shift on the Frank-Starling curve), resulting in decreased filling pressures and improved congestive symptoms. Although this fall in preload may be associated with a reduction in stroke volume, this effect is minimal in patients with elevated filling pressures. Pure venodilators similarly reduce filling pressures and congestive symptoms and have minimal effect on stroke volume. Arterial vasodilators and inotropic agents predominantly augment cardiac output and thereby improve organ perfusion; arterial vasodilators act indirectly through a reduction in vascular resistance, whereas inotropic agents directly increase contractility and stroke volume. Although the increased cardiac output seen with these agents may result in a fall in filling pressures, the effect may be relatively modest. As a result of the differential effects of these agents, many patients with heart failure attain the greatest benefit from combination therapy. Neurohormonal inhibitors may have mixed hemodynamic effects. The blockade of adrenergic tone (beta-blockers) and inhibition of the renin-angiotensin system (angiotensin-converting enzyme [ACE] inhibitors, angiotensin receptor blockers [ARBs]) results in vasodilation and augmented cardiac output. These agents may also decrease preload and reduce filling pressures through a reduction in neurohormonally mediated renal sodium and water retention.
Specific Agents
Diuretics
Diuretics have long played an important role in the symptomatic treatment of heart failure (Table 58-5). The induced diuresis and natriuresis reduce extracellular volume and ventricular filling pressures, thereby ameliorating congestive symptoms. This effect occurs without a significant decrease in cardiac output or systemic blood pressure unless excessive diuresis and intravascular volume depletion occur. Whereas diuretics are beneficial in controlling symptoms and improving exercise capacity in patients with heart failure, with the exception of spironolactone, eplerenone diuretic use has not resulted in a decrease in mortality.
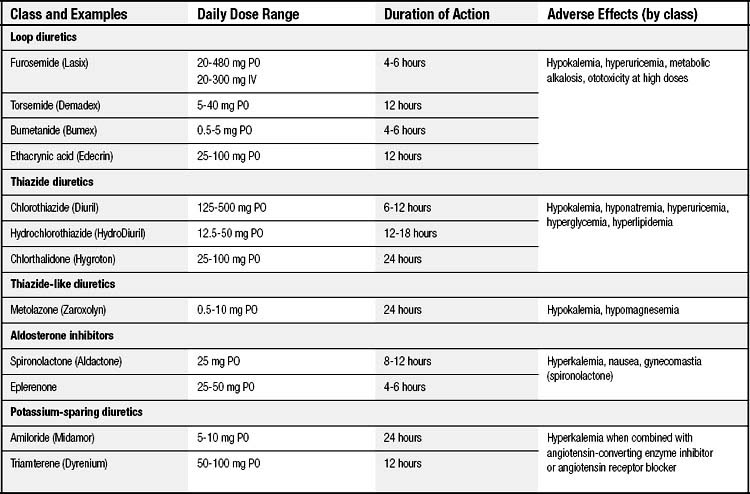
Furosemide is the loop diuretic most commonly used for the treatment of heart failure. For patients with mild to moderate congestive symptoms, it can be given orally at initial doses of 20 to 40 mg daily. Its bioavailability ranges from 40% to 70%, and gradual dose titration is frequently required. Furosemide has a relatively short half-life. Once renal tubular levels of the drug decline, avid sodium reabsorption occurs throughout the nephron, potentially limiting or preventing effective natriuresis. A twice-daily dosing regimen may therefore be required to produce adequate salt and water loss. In patients with more severe volume retention or decompensated heart failure, intravenous administration of furosemide (20 to 100 mg) may produce a more rapid and effective diuresis. The maximum intravenous dose is 300 mg; however, the risk of ototoxicity increases at such high doses. For patients who require frequent high doses of intravenous furosemide, a continuous infusion may produce a more effective diuresis and require a lower total daily dose. The infusion is usually started at 5 to 10 mg/hr and titrated as needed to obtain the desired effect. Bumetanide and torsemide have greater bioavailability than does furosemide (~80%) but have not demonstrated better efficacy and are significantly more expensive.
Vasodilators
Nitrovasodilators
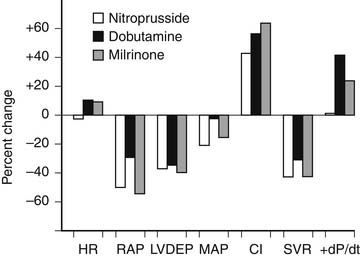
Nitroprusside is the sodium salt of nitric oxide and ferricyanide. It is a balanced vasodilator and produces both vasodilation and venodilation in both the systemic and pulmonary systems. These effects result in favorable hemodynamic changes, including a decrease in right atrial and pulmonary capillary wedge pressures (i.e., decreased preload), a reduction in pulmonary and systemic vascular resistance (i.e., decreased afterload), and an increase in stroke volume and cardiac output/index (Fig. 58-2). In contrast to other arterial vasodilators, nitroprusside does not cause a significant increase in heart rate, and its use is usually associated with a decrease in myocardial oxygen demand. Nitroprusside is useful for the management of heart failure associated with elevated filling pressures, low cardiac output, and high vascular resistance, as occurs in patients with decompensated systolic failure. It is also ideally suited for the management of heart failure associated with profound hypertension, acute mitral regurgitation, acute aortic insufficiency, or acute ventricular septal defect.
Hydralazine
Hydralazine therapy is initiated at a dose of 10 mg four times daily and titrated upward as blood pressure tolerates to a maximum dose of 100 mg four times daily. Organic nitrates are given concurrently (i.e., isosorbide dinitrate, 30 to 160 mg daily). Hydralazine-induced vasodilation is associated with a baroreceptor-mediated increase in sympathetic activity, resulting in a reflex tachycardia, increased ventricular contractility, increased renin activity, and fluid retention. In patients with underlying coronary artery disease, the arteriolar dilation may result in a coronary steal phenomenon and, combined with the tachycardia, may precipitate myocardial ischemia. Coadministration with β-adrenergic blocking agents may prevent this complication; nonetheless, hydralazine should be used with caution in patients with an ischemia cardiomyopathy. Other side effects occur more frequently and include headaches, flushing, palpitations, nausea, and dizziness. A lupus-like syndrome occurs in 5% to 10% of patients and may require discontinuation of the drug.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


