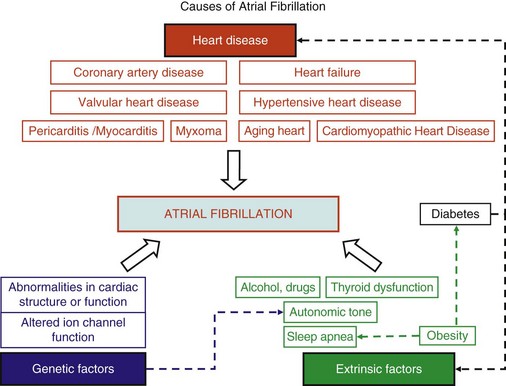
45 We will begin with an overview of the etiologic determinants of AF (Figure 45-1), then discuss briefly the principal mechanisms contributing to AF, and finally review the molecular basis of specific arrhythmia determinants. Most patients with AF have associated cardiac disease.1 Cardiac senescence is a major predisposing factor, largely mediated by structural remodeling, which causes fibrotic alterations and microconduction slowing, as well as atrial enlargement.2 Heart failure (HF), hypertensive heart disease, valvular disease, and ischemic heart disease are major contributors to AF occurrence. Less common conditions leading to AF include pericarditis, myocarditis, atrial tumors (primarily myxomas), and various cardiomyopathies. Knowledge of genetic determinants of AF has increased rapidly over the past 10 years.3,4 Disease-causing mutations have been established and the underlying pathophysiology studied.3 Monogenic forms have high penetrance and provide important mechanistic insights into AF mechanisms. Genome-wide association studies (GWASs) provide new insights into genetic variation–based population determinants of AF, while raising challenging pathophysiological issues.4 A variety of extracardiac conditions can affect AF occurrence. Heavy alcohol consumption promotes AF,5 and hyperthyroidism is a well-recognized factor.6 Obesity is increasingly recognized as an AF risk factor,7 with obstructive sleep apnea, often associated with obesity, also noted as an important contributor. Autonomic tone may set the conditions for AF initiation and maintenance. The AF-promoting properties of vagal activation are well known, and increasing evidence suggests an important role for combined sympathovagal discharge.8 AF can be maintained by rapid focal firing or by reentrant activity (Figure 45-2). To maintain AF, focal ectopic activity must be sustained, to produce rapid “driver” activity that is conducted heterogeneously to generate fibrillatory conduction and the irregular activity typical of AF.9,10 Focal activity can also be transient, producing isolated atrial extrasystoles or self-limited tachycardias. Transient focal activity can contribute to AF generation by acting as a trigger to initiate reentry in a vulnerable substrate. Clinical AF can be paroxysmal (self-terminating), persistent (terminating only with medical intervention), or permanent (continuing despite medical therapy). Repetitively firing focal ectopic drivers are believed to produce paroxysmal forms. Reentrant activity generates more persistent AF, tending to become resistant as the substrate evolves, as well as more fixed and irreversible.11,12 One factor contributing to evolution of the substrate is that AF induces remodeling, related to the rapid atrial rate and to other factors like cardiac dysfunction, neurohumoral changes, and consequences of atrial metabolic disturbances. The remodeling induced by chronic long-standing AF can involve both functional and structural changes that promote a transition toward complex reentrant mechanisms. Figure 45-2 Mechanistic Basis of Atrial Fibrillation (AF) and Associated Clinical Forms Focal ectopic activity may also result from afterdepolarizations, which are subdivided into early afterdepolarizations (arising before the end of phase 3) and delayed afterdepolarizations (DADs), which occur after full repolarization. Cell Ca2+ handling is crucial for normal contractility (Figure 45-3, A). DADs (Figure 45-3, B) are thought to be the most important cause of focal atrial ectopic firing. DADs are caused by a diastolic Ca2+ leak from the sarcoplasmic reticulum (SR) via SR Ca2+-release channels or ryanodine receptors (RyRs; RyR2 is the cardiac form). Systolic Ca2+ release through RyR2s causes cardiac contraction. Relaxation is mediated by diastolic Ca2+ removal from the cytosol into the SR by a Ca2+-uptake pump, the SR Ca2+-adenosine triphosphatase (ATPase; SERCA). RyR2s are sensitive to both cytosolic and intraluminal SR Ca2+ concentration; diastolic releases result when SR concentrations are excessive, or when RyR2s have an abnormally low threshold for Ca2+ release. Excess cytosolic Ca2+ is handled by the sarcolemmal Na+/Ca2+-exchanger (NCX), which moves three Na+ ions (charge +3) into the cell for each Ca2+ ion (charge +2) extruded into the extracellular space, generating net inward current (called transient inward current [Iti]) that depolarizes the cell, producing a DAD. When DADs reach threshold, they induce premature action potential firing (dashed line in Figure 45-3, B). Repeated DADs can generate focal atrial tachycardias. RyR2 function is regulated by channel phosphorylation: Hyperphosphorylation enhances RyR Ca2+ sensitivity and promotes DAD formation. Calsequestrin (CSQ) is the principal Ca2+-storage buffer of the SR. Inadequate CSQ function/expression increases free SR Ca2+ concentration and promotes diastolic RyR2 Ca2+ release.13 Figure 45-3 Cellular Ca2+ Handling and DADs Two conceptual frameworks for understanding the basis of functional reentry are shown in Figure 45-4 (top). The leading circle model (Figure 45-4, A) posits reentry around a central zone that is continuously activated by centripetal waves emanating from a reentering activation wave front. In this model, reentry establishes itself in a circuit for which the dimension equals the distance traveled during one refractory period (wavelength; refractory period times conduction velocity). When the wavelength is small because of slow conduction or brief refractoriness, multiple circuits can be accommodated in the atria and spontaneous self-termination is unlikely. In the spiral wave model (Figure 45-4, B), reentry is maintained by rotors established by tissue excitability properties (depending on both conduction and refractoriness conditions), which determine rotor rapidity, stability, and size (greater excitability generates smaller, more stable, and faster rotors). Anatomical obstacles or complexities can favor reentry by anchoring reentry circuits. Figure 45-4, C illustrates the effect of structural remodeling. Progressive atrial dilatation creates longer conduction pathways for reentry. Tissue fibrosis slows conduction and creates conduction barriers that favor the development of stable rotors and/or multiple simultaneous irregular reentry circuits that can sustain AF. In addition, fibroblast proliferation can promote arrhythmogenesis via cardiomyocyte-fibroblast interactions that alter AP properties and slow conduction. Figure 45-4 Determinants of Reentry Normal cell Ca2+ handling is crucial for cellular contraction and relaxation (see Figure 45-3, A; see also Chapter 16 of this book). Abnormal SR Ca2+ handling is typically seen in AF patients.14–19 Defective Ca2+ handling promotes spontaneous RyR2-mediated diastolic SR-Ca2+ releases in atrial cells from patients with chronic AF.14,15,18,19 Figure 45-5 summarizes the detailed molecular pathobiology of DAD-inducing diastolic RyR2 Ca2+ release. Protein kinase A (PKA) phosphorylation of RyR2 at Ser280817 and Ca2+ calmodulin–dependent kinase II (CaMKII) phosphorylation at Ser2814 are increased in dogs and goats with pacing-induced AF and in AF patients.14,19–21 CaMKII activity is normally autoinhibited. Ca2+-calmodulin binding removes autoinhibition, activating CaMKII and causing autophosphorylation that activates CaMKII and makes it Ca2+ independent. Similar activation may result from CaMKII oxidation. Changes in RyR2 phosphorylation state at PKA and CaMKII sites may result not only from changed kinase activity, but also from alterations in dephosphorylating enzyme phosphatases.22 These posttranslational alterations increase RyR2 Ca2+ sensitivity, enhancing channel open probability.14,17 Mice deficient in RyR2-inhibitory FK-505 binding protein 12.6, mice with gain-of-function mutations in RyR2, and mice with constitutively phosphorylated RyR2 channels at S2814 (S2814D mice) all exhibit increased susceptibility to pacing-induced AF in association with increased atrial cell SR Ca2+ leak and triggered activity.14,20,23,24 Angiotensin effects to promote AF may be due in part to oxidative stress acting via CaMKII oxidation on diastolic RyR2 Ca2+ release.25 RyR2 dysfunction can be induced by Ca2+ overload resulting from phospholamban hyperphosphorylation, which removes phospholamban inhibition of SERCA and enhances SR Ca2+ uptake.22 Phospholamban hyperphosphorylation can be produced by enhanced PKA or CaMKII activity, or by decreased phosphatase function. Reduced phosphatase function can be a consequence of increased activity of an inhibitory protein, I-1, typically caused by I-1 hyperphosphorylation.22 Figure 45-5 Molecular Basis of DAD-Inducing Diastolic Ca2+ Releases Increases in NCX expression and/or function are also commonly noted in AF,14,19,22,26 causing Iti resulting from any specific amount of diastolic SR Ca2+ leak to be larger in AF, likely contributing to the increased risk of DADs and triggered ectopic activity.14,27 Cardiac IP3 receptors (IP3R2) act as Ca2+-transporting pathways and can facilitate SR Ca2+ leak to promote arrhythmogenesis. IP3R2 expression is increased by atrial tachycardia remodeling (ATR) and is greater in atria than in ventricles.28 IP3R2-coupled amplification of atrial SR Ca2+-release events and related arrhythmogenesis may thus contribute to AF-related ectopic activity. Congestive heart failure (CHF) is a very important cause of AF. Focal drivers and triggered activity play a role in CHF-related AF.29 In experimental dilated cardiomyopathy, CHF increases SR Ca2+ load and reduces calsequestrin expression, thereby promoting spontaneous SR Ca2+ release.30 Coronary artery disease (CAD) is also an important risk factor for AF. Atrial ischemia promotes AF maintenance.31 In a dog model of chronic occlusive coronary artery disease affecting the atrium, frequent spontaneous atrial ectopy is associated with an increased incidence of atrial cardiomyocyte-triggered activity.31 Triggered activity is likely due to spontaneous SR Ca2+-release events and increased NCX function in cardiomyocytes from the ischemic border zone.31 Two AF-promoting genetic variants have been linked to DAD mechanisms: (1) a mutation of the gene encoding the adapter protein ankyrin-B (long QT syndrome-4 [LQTS4]), which causes multiple proteins to be poorly addressed to their membrane targets, altering Ca2+ handling and leading to DADs/triggered activity32,33; and (2) a predicted loss-of-function single-nucleotide polymorphism (SNP) of the SLN gene encoding the Ca2+-binding protein sarcolipin, which could increase SR Ca2+ load, thereby affecting DAD susceptibility.34 Beta-adrenoceptor activation phosphorylates RyR2, promoting diastolic SR Ca2+-release events.34 Conditions that directly cause DAD-promoting abnormalities in Ca2+ handling may require adrenergic stimulation to induce Ca2+ sparks and triggered activity.31 Spontaneous AF paroxysms occur in dog models of autonomic hyperinnervation,36 with sympathovagal discharge preceding AF paroxysms.37 Vagal activation promotes arrhythmogenesis by reducing APD, allowing afterdepolarizations induced by adrenergic stimulation to induce ectopic firing in pulmonary veins.38–40 AF41 and indeed all very rapid atrial tachyarrhythmias42 remodel atrial electrical properties to promote AF initiation and maintenance (ATR). A major AF-promoting component of ATR is refractory period reduction due to APD abbreviation (see Figure 45-4, B). Reduced depolarizing L-type Ca2+ current (ICa,L) and increased repolarizing inward-rectifier K+ currents underlie ATR-induced APD shortening.43–50 The molecular complex basis of ICa,L reduction seen in AF is illustrated in Figure 45-6, A. Rapid atrial activation induces Ca2+ loading, activating Ca2+-calmodulin/calcineurin/nuclear factor of activated T-lymphocyte (NFAT) signaling that causes downregulation of Cav1.2 α-subunit mRNA.51–53 Other contributors to ICa,L downregulation may include decreased expression of accessory β1-, β2a-, β2b-, β3-, and α2δ2-subunits45,54,55; Cav1.2 dephosphorylation via type 1 (PP1) and/or type 2A (PP2A) protein phosphatases22,45,56; and increased Cav1.2 α-subunit s-nitrosylation.57 MicroRNAs (miRNAs) are short RNA sequences that regulate cardiac gene expression by translational suppression and/or mRNA destabilization.58 They are centrally involved in cardiac remodeling and appear to play a major role in AF.58 Recent work implicates increased miRNA-328 in AF promotion due to ICa,L-downregulation mediated by inhibition of translation and mRNA destabilization.59 Finally, impaired Cav1.2 and Cav1.3 protein trafficking induced by a zinc-binding protein (Znt-1) and ankyrin B reduction, respectively, may contribute to ICa,L reduction in AF.33,60 Figure 45-6 Mechanisms of Ionic Current Remodeling in AF
The Molecular Pathophysiology of Atrial Fibrillation
Etiologic Determinants
Heart Disease
Genetic Determinants
Extracardiac Contributors
General Mechanisms and AF Forms
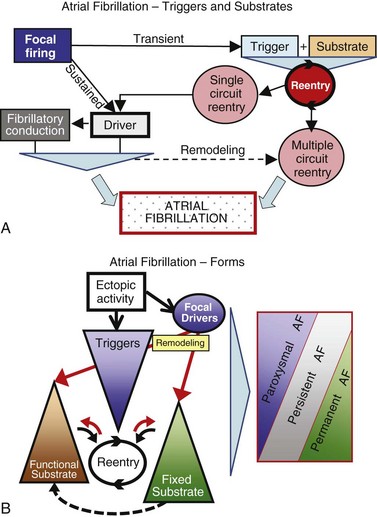
A, Focal firing usually results from local ectopic activity. Organized discrete reentrant activity and focal firing can maintain AF by producing regularly firing drivers that are conducted irregularly in the heterogeneous atrial substrate. B, Paroxysmal (self-terminating) AF is believed to result primarily from focal drivers, persistent AF from functional substrates, and permanent (nonterminating) AF from fixed fibrotic/anatomically remodeled substrates. However, overlap and partial contributions are indicated by the gray shading.
Focal Ectopic Activity
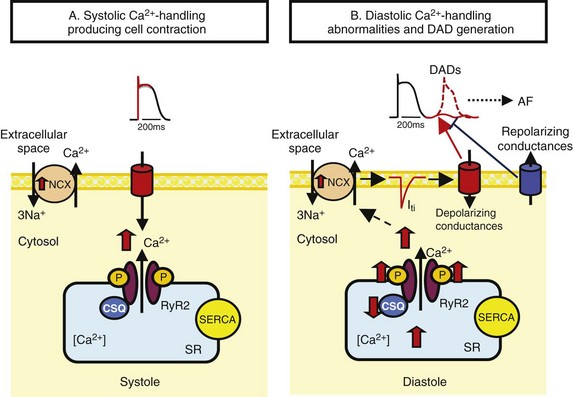
A, During the plateau phase of the action potential (AP), Ca2+ enters the cell via L-type Ca2+ channels. This Ca2+ binds to ryanodine-receptors (RyR2s), thereby triggering a much larger Ca2+ release from the sarcoplasmic reticulum (SR), which initiates cellular contraction. SR Ca2+ stores are maintained via Ca2+ pumping into the SR by the SR Ca2+–adenosine triphosphatase (ATPase; SERCA). B, Diastolic Ca2+-handling abnormalities underlie DADs. Spontaneous SR Ca2+ releases through RyR2 elevate cytosolic Ca2+, which is exchanged for extracellular Na+ by the Na+/Ca2+ exchanger (NCX), producing depolarizing current (transient inward-current [Iti]). Inappropriate diastolic RyR2 Ca2+ release is produced by RyR2 hyperphosphorylation, excess SR Ca2+, or decreased SR Ca2+ binding to calsequestrin (CSQ). Repolarizing conductances oppose Iti and suppress diastolic depolarization, so reduced diastolic K+ current can favor DADs P, Phosphate; Iti, transient inward current..
Reentry
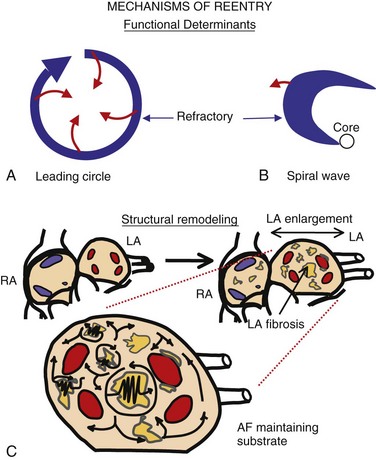
Top, Basic concepts of reentry. The leading circle model (A) posits reentry around a central zone that is continuously activated by centripetal waves emanating from the reentering activation wave front. The spiral wave model (B) describes reentry as a “rotor” established by tissue excitability properties. C, Structural remodeling (atrial enlargement and fibrosis, most typically affecting the left atrium [LA]) produces relatively fixed reentry substrates that reverse poorly if at all.
Molecular Control Mechanisms
Molecular Control of Cell Ca2+ Handling and DAD Generation
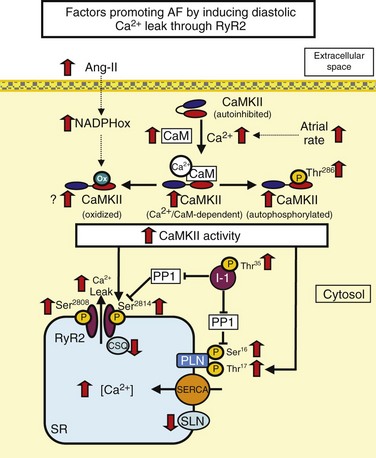
RyR dysfunction is caused by RyR hyperphosphorylation or excess Ca2+ loads. Phospholamban (PLN) inhibits SERCA. PLN hyperphosphorylation removes this inhibitory effect, enhances SERCA function, and can lead to Ca2+ overload. High atrial rate during AF enhances cellular Ca2+ entry. Increased cell Ca2+ promotes Ca2+/calmodulin (CaM) binding to Ca2+/calmodulin-dependent protein kinase II (CaMKII), disinhibiting the catalytic subunit. After CaMKII catalytic subunit activation, oxidation at Met281/282 or phosphorylation at Thr286 causes persistent CaMKII activity. Inhibitor-1 (I-1) suppresses protein phosphatase 1 (PP1) function in the SR and contributes to PLN and RyR phosphorylation. Ang-II, Angiotensin-II; NADPHox, Nicotinamide adenine dinucleotide phosphate oxidase; CaM, calcium/calmodulin; CaMKII, calcium-calmodulin dependent kinase type II; CSQ, calsequestrin; PLN, phospholamban; SERCA, sarcoplasmic reticulum calcium-ATPase; SLN, sarcolipin; RyR2, ryanodine receptor type 2.
Molecular Control of L-Type Ca2+ Current and Pathophysiology in AF
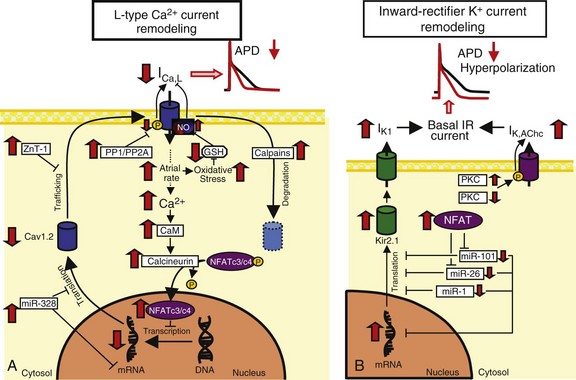
A, L-type Ca2+-current (ICa,L) downregulation. High atrial rates in AF enhance intracellular Ca2+ load and activating calcineurin via Ca2+/calmodulin (CaM) binding. Calcineurin dephosphorylates nuclear factor of activated T-lymphocytes (NFAT), allowing it to translocate into the nucleus and reduce mRNA levels of the ICa,L alpha subunit, Cav1.2. Breakdown of Cav1.2 protein by calpains might also contribute to reduced Cav1.2 protein expression. The protein zinc transporter 1 (ZnT-1) impairs Cav1.2 membrane trafficking and is upregulated in AF. Increased protein phosphatase (PP) activity dephosphorylates Cav1.2 phosphorylation and may decrease ICa,L. B, Inward-rectifier K+-current upregulation. Increased Kir2.1 subunit expression is caused by decreases in inhibitory microRNAs like miR-101, miR-26, and miR-1. Acetylcholine-regulated K+ current (IK,AChc) is increased because of increased membrane abundance of the stimulatory protein kinase C (PKC) isoform PKCε and decreased cellular expression of the inhibitory isoform PKCα. PP, Protein phosphatase; ZnT-1,![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The Molecular Pathophysiology of Atrial Fibrillation