Chapter 2 The Endothelium
In 1839, the German physiologist Theodor Schwann became the first to describe a “thin, but distinctly perceptible membrane” that he observed as part of the capillary vessel wall that separated circulating blood from tissue.1,2 The cellular monolayer that formed this membrane would later be named the endothelium; however, the term endothelium did not appear until 1865 when it was introduced by the Swiss anatomist Wilhelm His in his essay, “Die Häute und Höhlen des Körpers (The Membranes and Cavities of the Body).”2,3 Owing to its anatomical location, the endothelium was believed initially to be a passive receptacle for circulating blood, cells, and macromolecules. It is now known that the endothelium is a dynamic cellular structure, and its biological and functional properties extend beyond that of a physical anatomical boundary. In its totality, the endothelium comprises approximately 10 trillion (1013) cells with a surface area of 7 m2, weighs 1.0 to 1.8 kilograms, and contributes 1.4% to total body mass.4,5 Endothelium exists as a monolayer of cells that is present in all arteries, veins, capillaries, and the lymphatic system, and lies at the interface of the bloodstream or lymph and the vessel wall.
Homeostatic Functions of the Endothelium
The endothelium exhibits considerable regional heterogeneity that reflects its arterial or venous location in the vascular tree, as well as the specialized metabolic and functional demands of the underlying tissues.5–7 Despite this heterogeneity, there are basal homeostatic properties that are common to all endothelial cell (EC) populations, although some of these functions may achieve greater importance in selected vascular beds7 (Box 2-1).
 Box 2-1 Homeostatic Functions of the Endothelium
Box 2-1 Homeostatic Functions of the Endothelium
Maintenance of a Thromboresistant Surface and Regulation of Hemostasis
The endothelium was first recognized as a cellular structure that compartmentalizes circulating blood.4 As such, the endothelial luminal surface is exposed to cells and proteins in the bloodstream that possess prothrombotic and procoagulant activity and, when necessary, support hemostasis. Normal endothelium preserves blood fluidity by synthesizing and secreting factors that limit activation of the clotting cascade, inhibit platelet aggregation, and promote fibrinolysis.8 These include the cell surface–associated anticoagulant factors thrombomodulin, protein C, tissue factor pathway inhibitor (TFPI), and heparan sulfate proteoglycans (HSPG) that act in concert to limit coagulation at the luminal surface of the endothelium.8–10 For instance, thrombin-mediated activation of protein C is accelerated 104-fold by binding to thrombomodulin, Ca2 +, and the endothelial protein C receptor. Activated protein C (APC) engages circulating protein S, which is also synthesized and released by the endothelium, to inactivate factors Va and VIIIa proteolytically.8,11 Tissue factor pathway inhibitor is a Kunitz-type protease inhibitor that binds to and inhibits factor VIIa; about 80% of TFPI is bound to the endothelium via a glycosylphosphatidylinositol anchor and forms a quaternary complex with tissue factor – factor VIIa to diminish its procoagulant activity.12,13 Proteoglycan heparan sulfates that are present in the EC glycocalyx attain anticoagulant properties by catalyzing the association of the circulating serine protease inhibitor antithrombin III to factors Xa, IXa, and thrombin.8 Thus, these anticoagulant factors serve to limit activation and propagation of the clotting cascade at the endothelial luminal surface and thereby maintain vascular patency.
The endothelium also synthesizes and secretes tissue plasminogen activator (tPA) and the ecto-adenosine diphosphatase (ecto-ADPase) CD39 to promote fibrinolysis and inhibit platelet activation, respectively. Tissue plasminogen activator is produced and released into the bloodstream continuously, but unless tPA binds fibrin, it is cleared from the plasma within 15 minutes by the liver.8 Fibrin binding accelerates tPA amidolytic activity by increasing the catalytic efficiency for plasminogen activation and plasmin generation. Platelet activation at the endothelial luminal surface is inhibited by the actions of the ectonucleotidase CD39/NTPDase1 that hydrolyzes adenosine diphosphate (ADP), prostacyclin (PGI2), and nitric oxide (NO).8,14,15 Together these agents maintain an environment on the endothelial surface that is profibrinolytic and antithrombotic.
By contrast, in the setting of an acute vascular injury or trauma, the endothelium initiates a rapid and measured hemostatic response through regulated synthesis and release of tissue factor and von Willebrand factor (vWF). Tissue factor is a multidomain transmembrane glycoprotein (GP) that forms a complex with circulating factor VIIa to activate the coagulation cascade and generate thrombin.16 Tissue factor is expressed by vascular smooth muscle cells (VSMCs) and fibroblasts and by ECs only after activation. Tissue factor acquires its biological activity by phosphatidylserine exposure, dedimerization, decreased exposure to TFPI, or posttranslational modification(s) including disulfide bond formation between Cys186 and Cys209.17–19 This disulfide bond is important for tissue factor coagulation activity and may be reduced by protein disulfide isomerase, which is located on the EC surface.
The endothelium also synthesizes and stores vWF, a large polymeric GP that is expressed rapidly in response to injury. Propeptides and multimers of vWF are packaged in Weibel-Palade bodies that are unique to the endothelium. Once released, vWF multimers form elongated strings that retain platelets at sites of endothelial injury. Weibel-Palade bodies also contain P-selectin, angiopoietin-2, osteoprotegerin, the tetraspanin CD63/Lamp3, as well as cytokines, which are believed to be present as a result of incidental packaging.20 The stored pool of vWF may be mobilized quickly to the endothelial surface, where it binds to exposed collagen and participates in formation of a primary platelet hemostatic plug. The endothelium modulates this response further by regulating vWF size, and thereby its activity, through the action of the EC product ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type I motif, number 13).21 This protease cleaves released vWF at Tyr1605-Met1606 to generate smaller-sized polymers and decrease the propensity for platelet thrombus formation.21 Thus, the endothelium uses geographical separation of factors that regulate its anti- and prothrombotic functions to maintain blood fluidity yet allow for a hemostatic response to vascular injury.
Semipermeable Barrier and Transendothelial Transport Pathways
The endothelial monolayer serves as a size-selective semipermeable barrier that restricts the free bidirectional transit of water, macromolecules, and circulating or resident cells between the bloodstream and underlying vessel wall or tissues. Permeability function is determined in part by the architectural arrangement of the endothelial monolayer, as well as the activation of pathways that facilitate the transendothelial transport of fluids, molecules, and cells. This transport occurs via either transcellular pathways that involve vesicle formation, trafficking, and transcytosis, or by the loosening of interendothelial junctions and paracellular pathways22 (Fig. 2-1). Molecules that traverse the endothelium by paracellular pathways are size restricted to a radius of 3 nm or less, whereas those of larger diameter may be actively transported across the cell in vesicles.23 Although the diffusive flux of water occurs in ECs through aquaporin transmembrane water channels, the contribution of these channels to hydraulic conductivity and cellular permeability is limited.24
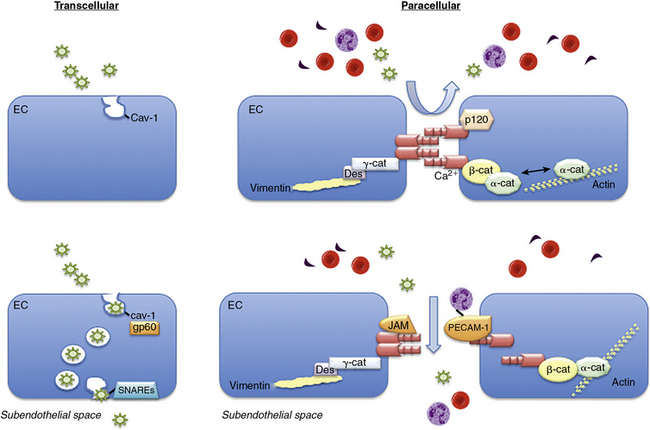
Figure 2-1 Transendothelial transport mechanisms.
(Adapted from Komarova Y, Malik AB: Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72:463–493, 2010.)
There is significant macrostructural heterogeneity of the endothelial monolayer that reflects the functional and metabolic requirements of the underlying tissue and has consequences for its permeability function. Endothelium may be arranged in either a continuous or discontinuous manner: continuous endothelium is either nonfenestrated or fenestrated.4–6
Continuous nonfenestrated endothelium forms a highly exclusive barrier and is found in the arterial and venous blood vessels of the heart, lung, skin, connective tissue, muscle, retina, spinal cord, brain, and mesentery.4–6 By contrast, continuous fenestrated endothelium is located in vessels that supply organs involved in filtration or with a high demand for transendothelial transport, including renal glomeruli, the ascending vasa recta and peritubular capillaries of the kidney, endocrine, and exocrine glands, intestinal villi, and the choroid plexus of the brain.4–6 These ECs are characterized by fenestrae, or transcellular pores, with a diameter of 50 to 80 nm that, in the majority of cells, has a 5- to 6-mm nonmembranous diaphragm across the pore opening.4–6,22 The distribution of these fenestrae may be polarized within the EC and allow for enhanced barrier size selectivity owing to the diaphragm.4–6
Discontinuous endothelium is found in the bone marrow, spleen, and liver sinusoids. This type of endothelial monolayer is notable for its large-diameter fenestrae (100-200 nm) with absent diaphragms and gaps, and a poorly organized underlying basement membrane that is permissive for transcellular flow of water and solutes as well as cellular trafficking.4–6
Transcellular and paracellular pathways are two distinct routes by which plasma proteins, solutes, and fluids traverse the endothelial monolayer. The transcellular pathway provides a receptor-mediated mechanism to transport albumin, lipids, and hormones across the endothelium.22,25,26 The paracellular pathway is dependent upon the structural integrity of adherens, tight, and gap junctions and allows fluids and solutes to permeate between ECs but restricts passage of large molecules.22,25,26 Although these pathways were believed to function independently, it is now recognized that they are interrelated and together modulate permeability under basal conditions.
The transcellular transport of albumin and albumin-bound macromolecules is initiated by albumin binding to gp60, or albondin, a 60-kDa albumin-binding protein located in flask-shaped caveolae that reside at the cell surface.27,28 These caveolae are cholesterol- and sphingolipid-rich structures that contain caveolin-1. Once activated, gp60 interacts with caveolin-1, followed by constriction of the caveolae neck and fission from the cell surface.29,30 These actions lead to formation of vesicles with a diameter of about 70 nm and vesicle transcytosis. Caveolae may contain as much as 15% to 20% of the cell volume, so they are capable of moving significant amounts of fluid across the cell through this mechanism.29,30 Once vesicles have detached from the membrane, they undergo vectorial transit to the abluminal membrane, where they dock and fuse with the plasma membrane by interacting with vesicle-associated and membrane-associated target soluble N-ethylmaleimide-sensitive factor attachment receptors (SNAREs).31 Once docked, the vesicles release their cargo to the interstitial space. Vesicles may traverse the cell as individual structures or cluster to form channel-like structures with a diameter of 80 to 200 nm that span the cell.5,6 Although transcellular vesicle trafficking is the predominant mechanism by which cells transport albumin, it is now appreciated that this pathway is not absolutely necessary for permeability function, owing to the compensatory capabilities of the paracellular pathway.
The junctions between ECs include the adherens, tight, and gap junctions; only the former two modulate permeability and comprise the paracellular pathway.32 Adherens junctions are normally impermeant to albumin and other large molecules and are the major determinant of endothelial barrier function and permeability. The expression of tight junctions, by contrast, is limited to the blood-brain or blood-retinal barriers where they restrict or prevent passage of small molecules (< 1 kDa) and some inorganic ions.22 Gap junctions are composed of connexins that form a channel between adjacent cells to enhance cell-cell communication and facilitate the transit of water, small molecules, and ions.22
Adherens junctions are critical for maintaining endothelial barrier functional integrity and are composed of complexes of vascular endothelial (VE)-cadherin and catenins. Vascular endothelial cadherin is a transmembrane GP with five extracellular repeats, a transmembrane segment, and a cytoplasmic tail. The external domains mediate the calcium-dependent hemophilic adhesion between VE-cadherin molecules expressed in adjacent cells.25,26,33 The cytoplasmic tail interacts with β-catenin, plakoglobin (γ-catenin), and p120 catenin to control the organization of VE-cadherin and the actin cytoskeleton at adherens junctions. The actin binding proteins α-actinin, annexin 2, formin-1, and eplin may further stabilize this interaction. Other proteins located in adherens junctions thought to provide stability include junctional adhesion molecules (JAMs) and platelet–EC adhesion molecule 1 (PECAM-1).22
Endothelial permeability may be increased or decreased through mechanisms that involve adherens junction remodeling or through interactions with the actin cytoskeleton.25,26,34 These events may occur rapidly, be transient or sustained, and are reversible. Most commonly, mediators that increase endothelial permeability either destabilize adherens junctions through phosphorylation, and thereby internalization, of VE-cadherin or by RhoA activation and actin cytoskeletal rearrangement to physically pull apart VE-cadherin molecules and adherens junctions, resulting in intercellular gaps.22 To counteract these effects, other mediators that attenuate permeability are present in the plasma or interstitial space. Fibroblast growth factor (FGF) stabilizes VE-cadherin by stabilizing VE-cadherin-gp120-catenin interaction. Sphingosine-1-phosphate, generated by breakdown of the membrane phospholipid sphingomyelin or released from activated platelets, also stabilizes adherens junctions. This effect occurs through activation of Rac1/Rap1/Cdc42 signaling and reorganization of the actin cytoskeleton, recycling of VE-cadherin to the cell surface, and (re)assembly of adherens junctions. The cytokine angiopoietin-1 stabilizes adherens junctions by inhibiting endocytosis of VE-cadherin.22,25,26,35,36
Endothelial tight junctions predominate in specialized vascular beds that require an impermeable barrier. These tight junctions are composed of the specific tight junction proteins occludin, claudins (3/5), and JAM-A.22,33,36,37 Occludin and claudins are membrane proteins that contain four transmembrane and two extracellular loop domains. The extracellular loop domains of these proteins bind similar domains on neighboring cells to seal the intercellular cleft and prevent permeability. Occludin, claudins, and JAM-A are also tethered to the actin cytoskeleton by α-catenin and zona occludens proteins (ZO-1, ZO-2).22 The ZO proteins also function as guanylyl kinases or scaffolding proteins and use PDZ and Sc homology 3 (SH3)-binding domains to recruit other signaling molecules. Connections between tight junctions and the actin cytoskeleton are stabilized further via the actin cross-linking proteins spectrin or filamen or by the accessory proteins cingulin and AF-6.22,36 In this manner, the junctions remain stabilized and sealed to limit or prevent transendothelial transport of fluids and molecules.
Regulation of Vascular Tone
Since the early seminal studies of Furchgott and Zawadski, it has been increasingly recognized that the endothelium regulates vascular tone via endothelium-derived factors that maintain a balance between vasoconstriction and vasodilation38,39 (Fig. 2-2). The endothelium produces both gaseous and peptide vasodilators, including NO, hydrogen sulfide, PGI2, and endothelium-derived hyperpolarizing factor (EDHF). The effects of these substances on vascular tone are counterbalanced by vasoconstrictors that are either synthesized or processed by the endothelium, such as thromboxane A2 TxA2, a product of arachidonic acid metabolism, and the peptides endothelin-1 (ET-1) and angiotensin II (Ang-II). The relative importance of these vasodilator or vasoconstrictor substances for maintaining vascular tone differs between vascular beds, with NO serving as the primary vasodilator in large conduit elastic vessels and non-NO mechanisms playing a greater role in the microcirculation.
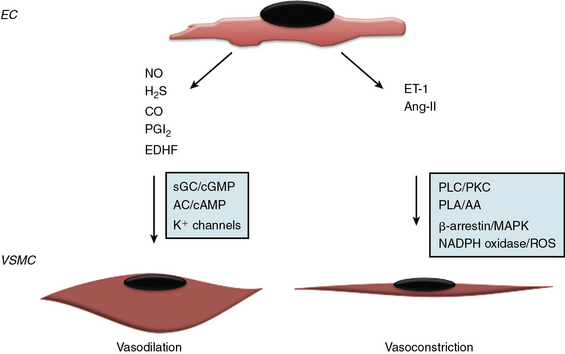
Nitric oxide is synthesized by three structurally similar NO synthase (NOS) isoenzymes: the constitutive enzyme identified in the endothelium (eNOS or NOS3) and neuronal cells (nNOS or NOS1) or the inducible enzyme (iNOS or NOS2) found in smooth muscle cells (SMCs), neutrophils, and macrophages following exposure to endotoxin or inflammatory cytokines.40–42 Nitric oxide is generated via a five-electron oxidation reaction of L-arginine to form L-citrulline and stoichiometric amounts of NO, and requires molecular oxygen and NADPH as co-substrates and flavin adenine dinucleotide, flavin mononucleotide, heme, and tetrahydrobiopterin as cofactors.43–45 In the endothelium, eNOS expression is up-regulated by a diverse array of stimuli including transforming growth factor (TGF)-β1, lysophosphatidylcholine, hydrogen peroxide, tumor necrosis factor (TNF)-α, oxidized low-density lipoprotein (LDL) cholesterol, laminar shear stress, and hypoxia, and is subject to both posttranscriptional and posttranslational modifications that influence activity, including phosphorylation, acetylation, palmitoylation and myristolation, as well as localization to caveolae.45 Once generated, NO diffuses into SMCs and reacts with the heme iron of guanylyl cyclase to increase cyclic guanosine monophosphate (cGMP) levels and promote vasodilation.42 Nitric oxide can also react with SH-containing molecules and proteins (e.g., peroxynitrite, N2O2) to generate S-nitrosothiols, a stable reservoir of bioavailable NO with recognized antiplatelet and vasodilator effects.46–48 In the presence of oxygen, NO can be oxidized to nitrite and nitrate, which are stable end-products of NO metabolism; nitrite serves as a vasodilator, predominantly in the pulmonary and cerebral circulations.48,49 In addition to vasodilator and antiplatelet effects, NO has other paracrine effects that include regulation of VSMC proliferation and migration, and leukocyte adhesion and activation.15
Hydrogen sulfide gas generated by the endothelium also possesses vasodilator properties. Hydrogen sulfide is membrane permeable and released as a byproduct of cysteine or homocysteine metabolism via the transulfuration/cystathionine-β-synthase and cystathionine-γ-lyase pathway or by the catabolism of cysteine via cysteine aminotransferase and 3-mercaptopyruvate sulfur transferase. Hydrogen sulfide–mediated vasodilation results from activation of KATP and transient receptor membrane channel currents.50–52
Prostacyclin is an eicosanoid generated by cyclooxygenase (COX) and arachidonic acid metabolism in the endothelium. It promotes vasodilation via adenylyl cyclase/cyclic adenosine monophosphate (cAMP) signal transduction pathways. Prostacyclin also induces smooth muscle relaxation by reducing cytoplasmic Ca2 + availability; decreases VSMC proliferation through a cAMP–peroxisome proliferator-activated receptor (PPAR)-γ-mediated mechanism, and limits inflammation by decreasing interleukin (IL)-1 and IL-6.53 Importantly, PGI2 has significant antiplatelet effects and by decreasing TxA2 levels, limits platelet aggregation. Because both COX-1 (constitutively expressed) and COX-2 (induced) contribute to basal PGI2 production, selective pharmacological inhibition of either isoform may result in diminished PGI2 levels, increased platelet aggregation, and impaired vasodilation.54
No single molecule has been identified as the vasodilator referred to as endothelium-derived hyperpolarizing factor, and the effects attributed to Endothelium-derived hyperpolarizing factor likely represent the composite actions of several agents that share a common mechanism. Endothelium-derived hyperpolarizing factor is an important vasodilator in the microcirculation and acts by opening K+ channels to allow for K+ efflux, hyperpolarization, and vascular smooth muscle relaxation. Candidate EDHFs include the 11, 12-epoxyeicosatrienoic acids and hydrogen peroxide.39,55–58
To counterbalance the effects of endothelium-derived vasodilators, the endothelium also synthesizes the vasoconstrictor ET-1 and metabolizes Ang I to Ang II. Endothelin-1, a 21-amino-acid peptide, is synthesized initially as inactive pre-proET-1 that is processed by endothelin-converting enzymes to its active form.59,60 Endothelin-1 binds to the G protein–coupled receptors (GPCRs) ETA and ETB: ECs express ETB, whereas SMCs express both receptors. Although activation of endothelial ETB increases NO production, concomitant activation of SMC ETA and ETB results in prolonged and long-lasting vasoconstriction that predominates.61
There is no evidence that ET-1 is stored for immediate early release in the endothelium, indicating that acute stimuli such as hypoxia, TGF-β, and shear stress that increase ET-1 production do so via a transcriptional mechanism; however, ET-1 and endothelin-converting enzyme are packaged in Weibel-Palade bodies.62 Endothelium also expresses angiotensin-converting enzyme (ACE) and, as such, modulates processing of Ang-I to the vasoconstrictor peptide Ang-II.63 Ang-II–stimulated activation of the Ang-I receptor results in vasoconstriction and SMC hypertrophy and proliferation, in part, by activating NADPH oxidase to increase reactive oxygen species (ROS) production.64–66 Vascular tone, therefore, is determined by the balance of vasodilator and vasoconstrictor substances synthesized or processed by the endothelium in response to stimuli: each vasoactive mediator may attain individual importance in a different vascular bed.
Regulating Response to Inflammatory and Immune Stimuli
The endothelium monitors circulating blood for foreign pathogens and participates in immunosurveillance by expressing Toll-like receptors (TLRs) 2, 3, and 4.67–69 These TLRs identify pathogen-associated molecular patterns that are common to bacterial cell wall proteins or viral deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) in the bloodstream. Once activated, TLRs elicit an inflammatory response through activation of nuclear factor (NF)-κB and generation of chemokines that promote transendothelial migration of leukocytes, have chemoattractant and mitogenic effects, and increase endothelial oxidant stress and apoptosis.67,68
The quiescent endothelium maintains its antiinflammatory phenotype through expression of cytokines with antiinflammatory properties and cytoprotective antioxidant enzymes that limit oxidant stress. The endothelium synthesizes TGF-β1, which inhibits synthesis of the proinflammatory cytokines monocyte chemotactic protein-1 (MCP-1) and IL-8; expression of the TNF-α receptor; NF-κB-mediated proinflammatory signaling; and leukocyte adherence to the luminal surface of the endothelium.70,71 Endothelium also expresses a wide array of antioxidant enzymes, including catalase, the superoxide dismutases, glutathione peroxidase-1, peroxiredoxins, and glucose-6-phosphate dehydrogenase.48 Through the actions of these antioxidant enzymes, ROS are reduced, and the redox environment remains stable. This homeostatic redox modulation also limits activation of ROS-stimulated transcription factors such as NF-κB, activator protein-1, specificity protein-1, and PPARs.48 The inflammatory phenotype of the endothelium is also influenced by other circulating or paracrine factors that have antioxidant or antiinflammatory properties, such as high-density lipoprotein (HDL) cholesterol, IL-4, IL-10, IL-13, and IL-1 receptor antagonist.5,6,72,73
The endothelium is capable of mounting a rapid inflammatory response that involves the actions of chemoattractant cytokines, or chemokines, and their associated receptors to facilitate interactions between leukocytes and the endothelium. Endothelial cells express the chemokine receptors CXCR4, CCR2, and CCR8 on the luminal or abluminal surface of cells.74 These receptors bind and transport chemokines to the opposite side of the cell to generate a chemoattractant gradient for inflammatory cell homing. Heparan sulfate (HS), which is present in the endothelial glycocalyx, may serve as a chemokine presenter and is necessary for the action of some chemokines such as CXCL8, CCL2, CCL4, and CCL5.75,76
Endothelial cells also express the Duffy antigen receptor for chemokines (DARC) that participates in chemokine transcytosis across cells. Duffy antigen receptor for chemokines is a member of the silent chemokine receptor family that has high homology to GPCRs and can bind a broad spectrum of inflammatory CC and CXC chemokines, including MCP-1, IL-8, and CCL5 or Regulated upon Activation, Normal T-cell Expressed, and Secreted (RANTES), but does not activate G-protein signaling.77–79 Exposure to chemokines, in turn, activates cellular signaling pathways that promote EC–leukocyte interactions; however, homing of leukocytes to tissues is mediated directly by cell surface adhesion molecules.
Endothelium expresses selectins and immunoglobulin (Ig)-like cell surface adhesion molecules that regulate endothelial-leukocyte interactions. P-selectin and E-selectin are lectin-like transmembrane GPs. These selectins mediate leukocyte adhesion through Ca2 +-dependent binding of their N-terminal C-type lectin-like domain with a sialyl-Lewis X capping structure ligand present on leukocytes.80–82 P-selectin is stored in Weibel-Palade bodies where it can be mobilized rapidly to the cell surface in response to thrombin, histamine, complement activation, ROS, and inflammatory cytokines. Cell surface expression of P-selectin is limited to minutes.80,82 By contrast, E-selectin requires de novo protein synthesis for its expression. E-selectin is expressed on the cell surface, but it may also be found in its biologically active form in serum as a result of proteolytic cleavage from the cell surface.5,81,82 These selectins bind the leukocyte ligands P-selectin glycoprotein ligand-1 (PSGL-1), E-selectin-ligand-1, and CD44, each of which appears to have a distinct function: PSGL1 is implicated in the initial tethering of leukocytes to the endothelium, E-selectin-ligand-1 converts transient initial tethers to slower and more stable rolling, and CD44 controls the speed of rolling.81,82
The Ig-like cell surface adhesion molecules expressed by the endothelium are intercellular adhesion molecule (ICAM)-1,ICAM-2, vascular cell adhesion molecule (VCAM)-1, and PECAM-1. Intercellular adhesion molecule-1 is expressed at low levels in the endothelium, but its expression is up-regulated several-fold by TNF-α or IL-1. Intercelluar adhesion molecule-1 is active when it exists as a dimer and is able to bind macrophage adhesion ligand-1 or lymphocyte function–associated antigen-1 on leukocytes to facilitate transendothelial migration.82,83 Clustering of ICAM-1 stimulates endothelial cytoskeletal rearrangements to form cuplike structures on the endothelial surface and remodel adherens junction complexes to enhance leukocyte transendothelial migration.82,84,85 Intercellular adhesion molecule-2, by contrast, is constitutively expressed at high levels by the endothelium, but its expression is down-regulated by inflammatory cytokines; however, ICAM-2 is believed to play a role in cytokine-stimulated migration of eosinophils and dendritic cells.86,87 Vascular cell adhesion molecule-1 is also up-regulated by inflammatory cytokines, binds to very late antigen-4 on leukocytes, and activates Rac-1 to increase NADPH oxidase activity and ROS production.82 PECAM-1 is expressed abundantly in adherens junctions and is involved in homophilic interaction between endothelial and leukocyte PECAM-1. This interaction stimulates targeted trafficking of segments of EC membrane to surround a leukocyte in preparation for transendothelial migration and typically occurs within 1 or 2 μm of an intact endothelial junction.82 The determination as to whether a leukocyte migrates paracellularly or transcellularly, therefore, appears to be dependent upon the relative tightness of endothelial junctions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


