CHAPTER 59 The Coronary Circulation
Dietary and Pharmacologic Management of Atherosclerosis
Accumulating experience from epidemiology, research trials, and clinical practice indicates that it is possible to prevent atherosclerotic disease with a high expectation of success and also to achieve stabilization or regression of established disease in many if not most patients. Since 1980, mortality due to coronary heart disease has declined approximately 50% in the United States.1 This may be attributed to number of factors—lower plasma cholesterol, improved treatment of hypertension, reduced smoking rates, intensive care for acute ischemia, and intervention by bypass surgery and angioplasty. Over the past 2 decades, more than 30 large randomized medical intervention trials have focused on clinical outcomes in patients with established coronary heart disease or with various levels of risk. The ability to alter the course of atherosclerotic disease by medical intervention has been proven beyond doubt. In smaller studies, usually employing combination therapy, event reductions exceeding 50% were suggested. Despite this progress, medical regimens for prevention and treatment of atherosclerosis remain underutilized. Thus, atherosclerotic vascular disease, which currently accounts for approximately one fourth of all deaths in the United States, is likely to remain a dominant clinical concern for decades to come. A cooperative interdisciplinary approach involving primary care physicians, midlevel providers, dietitians, cardiologists, endocrinologists, and surgeons will achieve the best results for individual patients. Physicians and allied health providers from a variety of subspecialties and specialties have gained expertise in lipid disorders and are able to provide consultative services on difficult patients. In the United States, clinical lipidologists may be located through the National Lipid Association (http://www.lipid.org), and comparable societies are forming throughout the world.
ATHEROSCLEROSIS
Lesion Development
Fatty streaks are early atherosclerotic lesions that first appear normally in teenagers in the coronaries and the aorta (Fig. 59-1). These lesions do not disrupt the endothelial lining and have no clinical consequence in themselves. Histologically, fatty streaks are usually found to consist of lipid-filled macrophages with lesser numbers of lipid-bearing smooth muscle cells. The macrophages are derived from circulating monocytes that adhere to and migrate across arterial endothelium—an effect mediated by cellular adhesion molecules upregulated in states of hypercholesterolemia and inflammation.
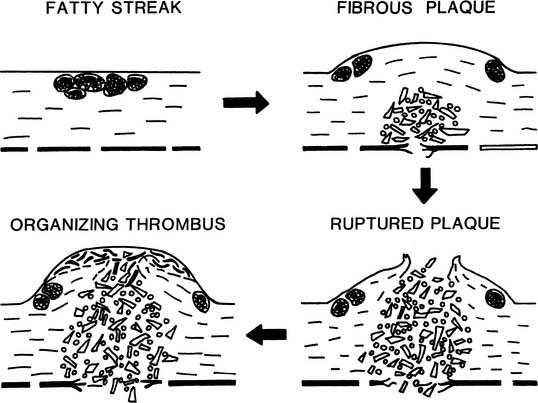
Beginning at about age 20, especially in men, raised lesions are found in the proximal coronaries, in the iliac arteries, and in the carotid bulb. Progression of raised atherosclerosis extends from the iliac arteries proximally into the abdominal and later thoracic aorta, and distally toward the femoral arteries. The raised character of the lesions results from the proliferation of smooth muscle cells and macrophages, and from elaboration of large amounts of fibrous tissue, especially collagen. Raised lesions with an intact intimal surface are called fibrous plaques. The majority of raised lesions possess a lipid-rich, hypocellular or acellular core region. Early development of the lipid-rich core has also been described in flat aortic lesions resembling fatty streaks.2 This finding is compatible with the hypothesis that most fibrous plaques are derived from preexisting fatty streaks (see Fig. 59-1).
As the fibrous plaque enlarges and begins to impinge on the arterial lumen, a compensatory enlargement of the entire artery occurs. This is probably a result of the physiologic regulation of arterial lumen diameter in response to blood velocity and shear rate. However, the compensatory enlargement eventually fails, perhaps when the collagenous lesion extends circumferentially almost all the way around the artery, leaving little normal wall to respond to blood flow.3
The most hazardous component of the atherosclerotic lesion is the lipid-rich core, which expands and erodes the fibrous cap of the lesion with time. When the fibrous cap finally ruptures, blood dissects rapidly into the core, and the thrombogenic contents of the core erupt into the vessel lumen, causing partial or complete thrombosis of the artery within a few minutes.4
Opportunities for Lesion Regression and Stabilization
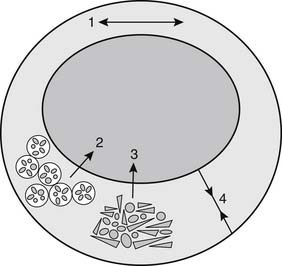
Atherosclerotic regression is often conceived of as a diminution in the size of an atherosclerotic lesion that results in decreased stenosis. Such regression may occur, but other, more readily achievable goals should also be considered (Fig. 59-2). For example, hypercholesterolemia can interfere with normal vasorelaxant function of endothelium, and decreases in low-density-lipid (LDL) cholesterol can normalize endothelial-dependent relaxation.5 The presence of macrophages in the fibrous cap of an atherosclerotic lesion predisposes the lesion to rupture.6 Lipid lowering can cause relatively rapid regression of foam cells, which appears to stabilize the fibrous cap, prevent rupture, and forestall atherothrombotic events.
LIPOPROTEIN METABOLISM
Lipoproteins and Apolipoproteins
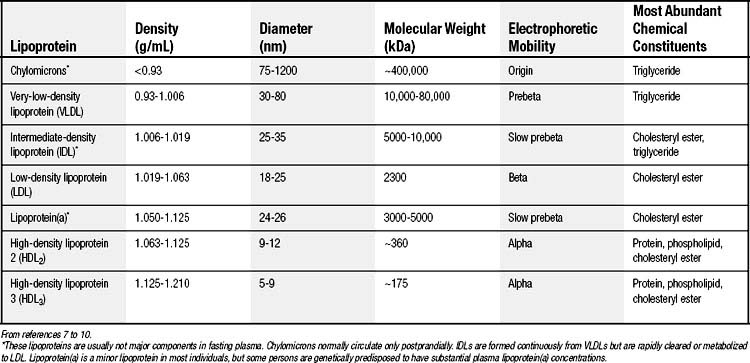
Human plasma lipoproteins function to transport triglyceride and cholesterol/cholesteryl ester in plasma. They are spherical particles containing 100 to several million lipid molecules combined with one or more protein molecules. The oily core of lipoproteins is composed of triglycerides and cholesteryl esters, and lipoprotein classification is based on which of these species is predominant (Table 59-1).7–10 The surface layer of lipoproteins is composed of phospholipids, free (unesterified) cholesterol, and protein.
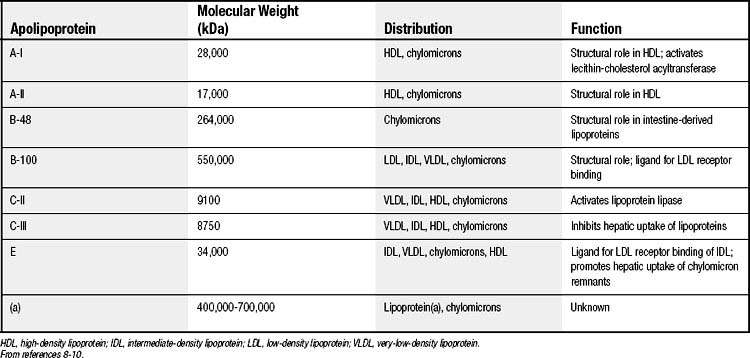
The protein components, termed apolipoproteins, bear specific binding sites for lipoprotein receptors or for enzymes involved in lipoprotein metabolism and thus target the lipoproteins for tissue uptake or lipid delivery (Table 59-2).8–10 All normal human lipoproteins contain either apolipoprotein A-I (apoA-I) or apolipoprotein B (apoB), or in the case of chylomicrons, both. Thus apoA-I and apoB appear to determine lipoprotein formation and structure as well as metabolic targeting. High-density lipoproteins (HDLs) characteristically contain apoA-I, whereas all lipoproteins of larger size and lower density contain apoB.
Lipoprotein Pathways
The metabolic pathways of apoB-containing lipoproteins originating in the liver are shown in Figure 59-3. A single molecule of apoB is incorporated into very-low-density lipoprotein (VLDL) before secretion from the liver, and this apoB molecule remains with the lipoprotein throughout all of its subsequent transformations, until the entire lipoprotein is taken up and degraded by a cell. VLDLs, which are triglyceride rich, encounter lipoprotein lipase and deliver fatty acids to tissues in exactly the same manner as chylomicrons. As shown in Figure 59-3, some of the VLDL remnants (also known as intermediate-density lipoproteins, or IDL) are processed within the hepatic microcirculation to become low-density lipoproteins, the major cholesterol-carrying particles in plasma. Physiologic removal of LDL from plasma depends on the presence of LDL receptor molecules found on the surface of hepatocytes and peripheral cells.
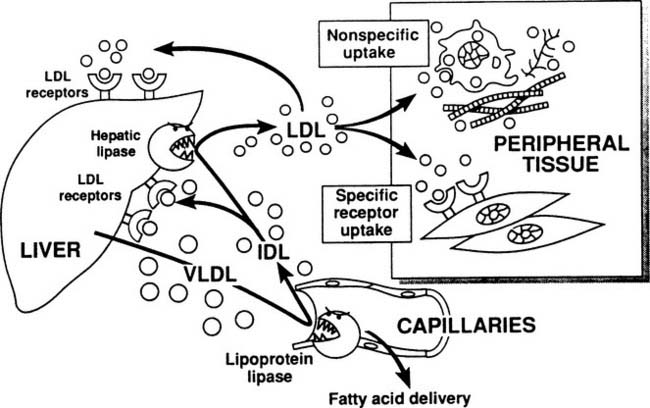
Triglyceride Metabolism
The level of triglycerides in plasma depends on the balance of their entry in the form of VLDL and chylomicrons, and their exit via the action of lipoprotein lipase. Deficiency of lipoprotein lipase action can result from partial or complete genetic defects in the enzyme itself or in its cofactor, apoC-II, or from suppression by diabetes mellitus or ethanol abuse. Any of these factors can raise plasma triglyceride levels greatly. The most important regulator of VLDL production is body fat. Intracellular lipase activity in adipose tissue causes the release of fatty acids, which circulate as plasma free fatty acids to the liver. The liver re-esterifies the fatty acids, making triglyceride for secretion in VLDL. Thus excess body fat stimulates fatty acid–triglyceride cycling between liver and the periphery, marked clinically by high plasma levels of triglyceride in VLDL. Improved caloric balance—that is, reduced dietary calories or increased caloric expenditure with exercise—is the only way to reduce body fat and counter this stimulus to high plasma triglyceride. Another stimulus to high plasma triglyceride is a high percentage of carbohydrates in the diet.11
Cholesterol Metabolism
Between 30% and 60% of dietary cholesterol is absorbed, and it reaches the liver largely via chylomicron remnants. A more important source of cholesterol, however, is synthesis in the liver and other body tissues from acetyl coenzyme A (CoA). A rate-limiting step in synthesis is catalyzed by 3-hydroxy-3-methylglutaryl CoA reductase, which is potently inhibited by statins, a category of cholesterol-lowering drugs. The liver is also the site of exit for most of the cholesterol leaving the body’s metabolic pools. Cholesterol cannot be broken down by mammalian cells to simple metabolic end-products such as carbon dioxide. The liver receives cholesterol by uptake of chylomicron and VLDL remnants, by uptake of a large percentage of LDL via the classic receptor expressed on hepatocytes, and by selective uptake of cholesteryl ester from HDL (see paragraph on reverse cholesterol transport, later). The liver converts cholesterol into bile acids; biliary secretion of these and of cholesterol itself account for the bulk of cholesterol removal from the body. More than 95% of bile acids are ordinarily reabsorbed in the terminal ileum.12 Oral ingestion of nonabsorbable bile acid binders (cholestyramine, colestipol, colesevelam) can lead to far greater net excretion in the stool and thus effectively reduce body cholesterol via bile acid conversion. Inhibition of intestinal absorption of cholesterol itself (e.g., by ezetimibe or by plant sterol/stanol esters) also lowers cholesterol in liver and in plasma LDL. These strategies are particularly effective when combined with agents that block a compensatory increase in cholesterol synthesis. The profound role of the liver in cholesterol metabolism is the reason that liver transplantation can reverse the extreme hypercholesterolemia that occurs in patients with homozygous deficiency of LDL receptors.
Heterozygous LDL receptor deficiency leads to expression of half the normal numbers of functional cell surface receptors and to approximately twice the normal plasma LDL levels. This highly atherogenic condition occurs in 1 of 500 persons worldwide.13 In a similar but much less frequent condition, heterozygous familial defective apoB, the binding site for the receptor is genetically dysfunctional on half of the circulating apoB molecules.14 The most common genetic hyperlipidemia that leads to coronary artery disease is neither of these, however, but is familial combined hyperlipidemia, a multifactorial or polygenic condition characterized by oversecretion of VLDL. Among different members of a family or even in a single individual at different times, either LDL (plasma cholesterol) or VLDL (plasma triglyceride) or both may be elevated.15
Peripheral cells can incorporate cholesterol by endocytic uptake of LDL via LDL receptors. Macrophages, which have the function of clearing proteinaceous debris from body tissue, express other receptors such as CD36 and scavenger receptor A that bind chemically altered, oxidized, or aggregated LDL.16
Reverse cholesterol transport—that is, the movement of cholesterol from peripheral tissues back to the liver—has been clarified by the discovery of key facilitating molecules. Cholesterol loading of macrophages induces expression of the adenosine triphosphate–binding cassette A1 (ABC-A1) transporter, which transfers unesterified cholesterol to lipid-poor apoA-I and forms nascent HDL.17 In disc-shaped nascent HDL, the enzyme lecithin-cholesterol acyltransferase (LCAT) functions to esterify cholesterol and thereby creates the oily lipid core of spherical HDL particles (which constitute almost all of the HDL in plasma). HDL can transport cholesterol to the liver by binding temporarily to scavenger receptor B1 on hepatocytes, which selectively removes cholesteryl ester from HDL and then releases the HDL particle for another round of reverse cholesterol transport. An alternative pathway for HDL cholesteryl ester is transfer via cholesteryl ester transfer protein (CETP) to LDL and VLDL, which are then taken up in the liver.16 Overall, the system is complex, and its regulation remains incompletely understood. For example, deficiency or inhibition of CETP can raise HDL levels, but it remains unproved whether the effect is antiatherogenic.18
Role of Lipoproteins in Atherogenesis
The concentration of LDL in the arterial intima is approximately equal to the plasma LDL concentration, whereas interstitial fluid elsewhere in the body appears to have a concentration one-tenth that in the plasma. In other tissues, lymph vessels act as sumps to carry away excess tissue macromolecular species such as LDL, but lymph vessels are absent from arterial intima, perhaps because of high hydrostatic pressure in the intima. Furthermore, the tunica media is highly impermeable to LDL, so that slow diffusion or convection of LDL across the arterial endothelium leads to high LDL concentrations in the intima. The relatively high intimal LDL concentration probably explains why, with aging, pathologic deposits of cholesterol develop regularly only in the arterial intima and nowhere else.19
Intimal lipid deposits are found both intracellularly and extracellularly at various stages of atherosclerosis. In addition to cholesteryl ester in superficially located foam cells, extracellular lipid deposits rich in unesterified cholesterol are found in the deep intimal core of small fibrous plaques. The formation of these deposits is unexplained; their importance is emphasized by an association with the disappearance of cells and a weakening of tissue in the developing core.20
Oxidation of LDL may help to explain a number of pathogenic processes in atherosclerosis. Certain lipid oxidation products are inflammatory mediators, and mildly oxidized LDL stimulates endothelial cells to express cell surface adhesion molecules for attachment of monocytes. However, large clinical trials have not supported a role for antioxidants in the prevention of clinical atherosclerotic events (see Table 59-6).
Lipoprotein(a)
Lipoprotein(a) is an atherogenic lipoprotein whose role in lipid metabolism remains somewhat mysterious. It is essentially an LDL with an added large glycopeptide, apolipoprotein(a), attached to apoB via a cysteine-cysteine disulfide bond. Lipoprotein(a) particles self-aggregate easily, perhaps favoring their deposition in the arterial intima. The amino acid sequence of apolipoprotein(a) includes multiple repeats of a “kringle” sequence found in plasminogen and in tissue plasminogen activator. The kringle (named after a Danish pastry) fosters the attachment of these fibrinolytic enzymes to sites of clot formation. Lipoprotein(a) has been shown to interfere competitively with binding of the enzymes and thus with fibrinolysis. Clinically, a strong positive correlation has been found between lipoprotein(a) levels and various manifestations of atherosclerosis, including myocardial infarction and stroke. Lipoprotein(a) levels show a strong inheritance pattern and are largely unaffected by diet and medications, except that niacin and oral estrogen can give 20% to 30% reductions.21
The Metabolic Syndrome of Insulin Resistance and Cardiovascular Risk
The metabolic syndrome includes (1) disordered metabolism of body fuels—glucose and fat—featuring insulin resistance and abdominal obesity, (2) atherogenic dyslipidemia with high triglyceride, low HDL cholesterol (HDLC), and small dense LDL, (3) hypertension, and (4) abnormalities of coagulation and inflammation adding to high cardiovascular risk. In the industrialized world, this syndrome makes a greater contribution to atherosclerotic disease than any other condition. By criteria of the National Cholesterol Education Program (Table 59-3), more than 40% of U.S. adults older than 60 have the metabolic syndrome.22
Table 59–3 Clinical Identification∗ of the Metabolic Syndrome
| Risk Factor | Defining Level |
|---|---|
| Abdominal obesity | Waist circumference |
| Men | >40 inches (>102 cm) |
| Women | >35 inches (>88 cm) |
| Triglyceride | ≥150 mg/dL |
| High-density lipoprotein cholesterol | |
| Men | <40 mg/dL |
| Women | <50 mg/dL |
| Blood pressure | ≥130/≥85 mm Hg |
| Fasting glucose | ≥110 mg/dL |
∗ Diagnosis of the metabolic syndrome requires any three of these risk factors.
In the metabolic syndrome, insulin resistance occurs in the phosphatidylinositol (PI) 3-kinase pathway of insulin signaling in cells, but not in the mitogen-activated protein (MAP) kinase pathway. The PI 3-kinase pathway is responsible for metabolic actions such as glucose uptake in peripheral cells and suppression of fatty acid release from adipocytes. The MAP kinase pathway, which may be overstimulated, leads to potentially atherogenic, mitogenic and proinflammatory effects.23,24
Fat overload and spillover can promote the metabolic syndrome.25 Fat is the body’s chief way of storing energy for future muscular activity. However, there is a finite capacity for the preferred site of energy storage in small peripheral adipocytes. Chronic energy imbalance—that is, too many calories in the diet or too few expended in exercise, or both—leads to spillover storage of triglyceride in skeletal myocytes, in hepatocytes, in excessive visceral adipocytes, and in abnormally large peripheral adipocytes. Excessive triglyceride in these sites leads to insulin resistance.26 Patients with lipodystrophy, either genetically based or caused by protease inhibitor therapy for human immunodeficiency virus, have a striking lack of small peripheral adipocytes.27 They develop central adiposity, hypertriglyceridemia, fatty liver, and insulin resistance or diabetes in an exaggerated form of the metabolic syndrome.
The dyslipidemic triad consists of high plasma triglyceride, low HDLC, and small dense LDL. In the metabolic syndrome, insulin resistance causes unsuppressed lipolysis of adipocyte triglyceride, which then leads excessive fatty acid delivery to the liver. The liver re-esterifies the fatty acids and exports them as VLDL triglyceride. Cholesteryl ester transfer protein mediates the exchange of VLDL triglyceride for cholesteryl ester in LDL and HDL. LDL and HDL thereby become enriched in triglyceride at the expense of core cholesteryl ester. The triglyceride-enriched LDL and HDL are subject to triglyceride hydrolysis by lipases that remove core lipids and make the lipoprotein particles smaller. Small dense LDLs appear to be more atherogenic than normal LDLs. Small dense HDLs are removed more rapidly from the circulation than normal HDLs, leading to reduced HDLC levels—another atherogenic situation.28
The adipocyte is now recognized as a source for multiple bioactive peptides, including angiotensinogen, tumor necrosis factor-alpha (TNF-α), leptin, and plasminogen activator inhibitor-1 (PAI-1).29 Overproduction of angiotensinogen may help to explain the link between obesity and hypertension. PAI-1, which inhibits fibrinolysis, is itself a cardiovascular risk factor. The liver appears to overproduce C-reactive protein, an extraordinarily strong marker of cardiovascular risk, in response to inflammatory cytokines from excessive fat deposits.30 Interestingly, low levels of cardiovascular fitness are independently associated with high C-reactive protein levels—another example of the relationship between energy metabolism and cardiovascular risk.31
CLINICAL MANAGEMENT
Major Clinical Trials
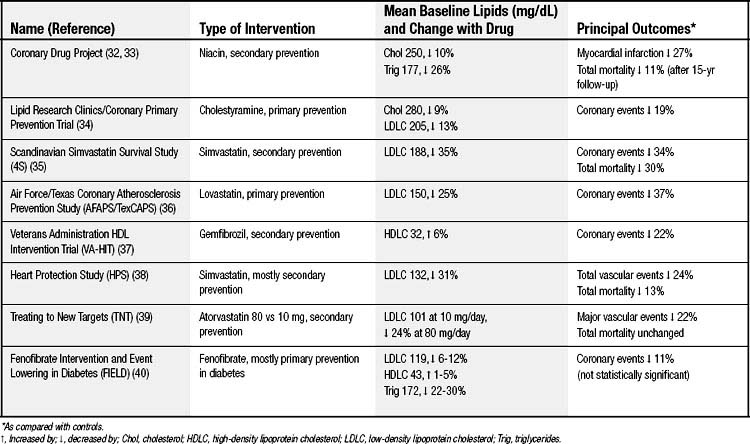
Medical treatment aimed at atherosclerosis can be classified as primary or secondary prevention depending on whether a person has previously presented clinically with symptomatic atherosclerotic disease such as angina or infarction. Tables 59-4, 59-5, and 59-6 show some of the clinical trials that shape our understanding of therapeutic efficacy.32–50 The risk for hard clinical endpoints, including myocardial infarction, coronary death, and stroke, was reduced sharply in several of these studies. Statins, or inhibitors of hydroxymethylglutaryl CoA reductase, have been found to be particularly effective, and some dietary interventions have also been surprisingly effective. Large trials such as these are expensive, and the temptation is to rely on smaller trials with soft clinical endpoints, such as hospitalizations or revascularizations for angina, or with anatomic endpoints, such as carotid intima–media thickness measurements. Although the smaller trials can provide some direction, they are inherently unreliable because of unaccounted covariance, and ultimately hard clinical outcomes must be ascertained.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


