CHAPTER 119 Tetralogy of Fallot with Pulmonary Stenosis
HISTORY
In 1888, Etienne-Louis Arthur Fallot described the four features of the congenital cardiac anomaly that bears his name: (1) ventricular septal defect (VSD), (2) infundibular pulmonic stenosis, (3) right ventricular hypertrophy, and (4) dextroposition of the aorta.1 More than 50 years later, the first successful surgical therapy for tetralogy of Fallot (TOF) was reported by Blalock and Taussig in 1945 after they performed a palliative subclavian-to-pulmonary-artery shunt.2 Waterston3 and Potts and colleagues4 also devised systemic-to-pulmonary arterial shunts with anastomoses between the descending aorta and left pulmonary artery or between the ascending aorta and right pulmonary artery, respectively. The first successful repair of this condition was performed by Lillehei and Varco, at the University of Minnesota, in 1954 using “controlled cross circulation,” with another patient serving as the oxygenator and blood reservoir.5,6 Kirklin reported the first repair of TOF using a pump oxygenator at the Mayo Clinic in 1955.7 Ross, Kirklin, Warden, Barrett-Boyes, Castaneda, and others made important contributions to the surgical management of TOF, including timing of operative repair, indications for the use of a transanular patch to relieve right ventricular outflow tract (RVOT) obstruction, and the use of grafts to reconstruct the RVOT.8–12
ANATOMY AND PHYSIOLOGY
Infundibular Septum
The four anatomic components originally described for TOF can be attributed directly to, or seen as a consequence of, a single anatomic abnormality—namely, anterior and leftward displacement of the infundibular or infundibular septum.13 This anomaly of the infundibular septum variably narrows the RVOT, leading to subpulmonic obstruction that may be complete (atresia). Displacement of the infundibular septum away from the anterior and posterior limbs of the trabecula septomarginalis results in the typical anterior malalignment VSD (see Ventricular Septal Defect, later). Because the aortic valve is located directly behind the infundibular septum, with anterior displacement of the infundibular septum, the aortic valve overrides the interventricular septum and attains some degree of association with the right ventricular cavity (Figs. 119-1 and 119-2). Right ventricular hypertrophy is a consequence of equalization of pressures in the right and left ventricles by virtue of an unrestrictive VSD.
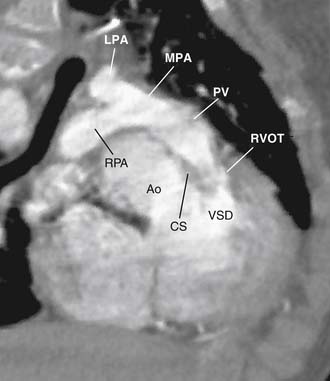
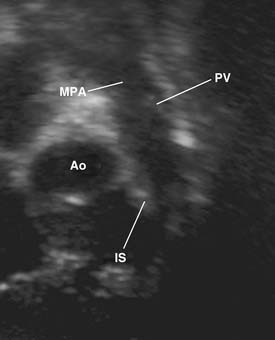
Figure 119–2 Long-axis echocardiographic view of the right ventricular outflow tract (the orientation is similar to that in Fig. 119-1). Ao, aorta; IS, infundibular septum; MPA, main pulmonary artery; PV, pulmonary valve.
Pulmonary Valve and Pulmonary Valve Anulus
The pulmonary valve is stenotic in 75% of cases; in half to two thirds of cases, the valve is bicuspid.14–16 Valve stenosis is usually caused by hypoplasia and fusion of bicuspid leaflets, supravalvar tethering, or a combination of these factors. Tethering of the leaflets often distorts the main pulmonary artery at the sinotubular junction, forming a ridge that may be obstructive, producing supravalvar stenosis. The leaflets themselves are often thickened with myxomatous excrescences, and when they are most severely affected, the leaflets may have a rigid, cartilaginous appearance (Fig. 119-3). The pulmonary valve anulus is invariably smaller than the aorta (the opposite of normal); however, it is not necessarily significantly obstructive. In symptomatic infants, the pulmonary valve anulus usually does contribute to multilevel RVOT obstruction.
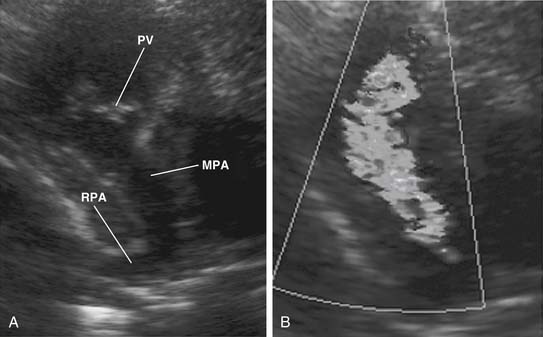
Main Pulmonary Artery and Branch Pulmonary Arteries
The main pulmonary artery is usually somewhat diffusely small and is often short, with a posterior angulation above the valve. The narrowest portion of the main pulmonary artery is often at the sinotubular junction because of the previously mentioned valvar narrowing at that level (Figs. 119-4 and 119-5).
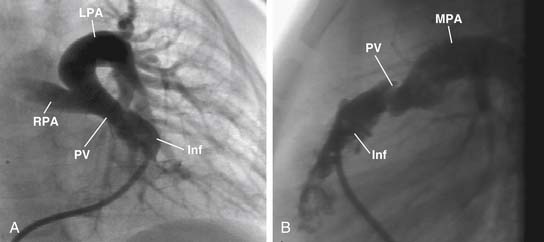
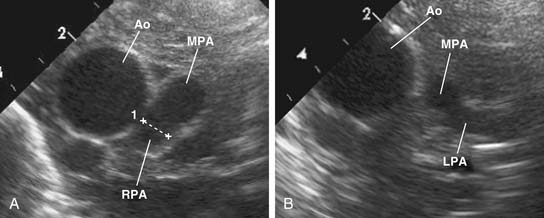
The left pulmonary artery usually appears to be a direct continuation of the main pulmonary artery, with the right pulmonary artery branching at nearly a right angle; however, important stenoses of the branch pulmonary arteries occur relatively infrequently. Analysis performed at the Green Lane Hospital in Auckland, New Zealand, demonstrated that branch pulmonary artery abnormalities occurred in only 10% of cases.15 Bilateral branch pulmonary artery stenosis made up approximately half of these cases, with juxtaductal stenosis of the left pulmonary artery occurring next most frequently. Beyond their origins, the branch pulmonary arteries themselves are not abnormally small on average, but in the pulmonary parenchyma, the sizes of the intra-acinar arteries and the alveoli are reduced, as are the lung volumes.17,18
Ventricular Septal Defect
As noted, the classic VSD typically seen in TOF is an anterior malalignment VSD arising because of anterior displacement of the infundibular septum away from the trabecula septomarginalis. Because of its close association with the posterior infundibular septum, the aortic valve overrides the interventricular septum and associates with the anterior and superior extent of the defect. The membranous septum is usually absent or attenuated, and the defect usually extends under the septal leaflet of the tricuspid valve and at times into the inlet portion of the interventricular septum. The superior margin of the VSD usually consists of fibrous tissue composed of the anteroseptal commissure of the tricuspid valve and the right fibrous trigone at the nadir of the right coronary cusp of the aortic valve. The posterior limb of the trabecula septomarginalis forms the posteroinferior border of the VSD, which buries the conducting bundle under a muscular rim and gives rise to the papillary muscle of the conus, which provides a marker for the course of the right bundle branch of the conducting bundle. In about 25% of cases, the ventriculo-infundibular fold extends inferiorly, separating the VSD from the tricuspid anulus, and with the posterior limb of the trabecula septomarginalis it forms an additional muscle layer over the conducting bundle.14,19 Anteriorly, the muscular margin of the VSD is composed of the anterior limb of the trabecula septomarginalis.
The VSD is almost always large and unrestrictive, with a diameter equal to or greater than that of the aortic anulus; the direction of shunting at the level of the defect therefore depends on the degree of RVOT obstruction (Figs. 119-6, 119-7, and 119-8). Typically, shunting is bidirectional but with a significant right-to-left component throughout much of the cardiac cycle. Systemic arterial desaturation occurs because of the limitation of blood flow into the pulmonary arteries as well as intracardiac right-to-left shunting, with the extent of clinically evident cyanosis being inversely proportional to the amount of pulmonary blood flow. Patients with a good-sized pulmonary valve anulus and relatively mild infundibular stenosis may have systemic arterial saturations that approach normal, or they may even present with congestive heart failure. On the other hand, children with severe obstruction to pulmonary blood flow may be severely cyanotic in the neonatal period. Anatomically, these children typically have multilevel RVOT obstruction with narrowing of the infundibulum, hypoplasia of the pulmonary valve anulus, a small main pulmonary artery, and, possibly, small branch pulmonary arteries.
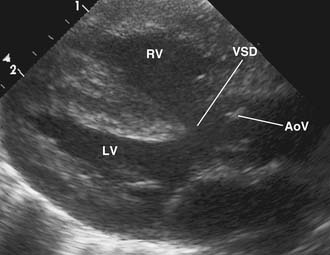
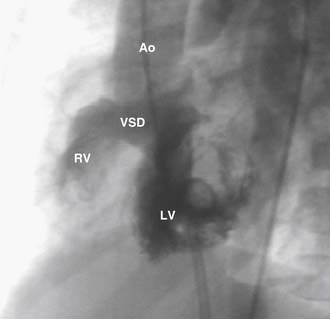
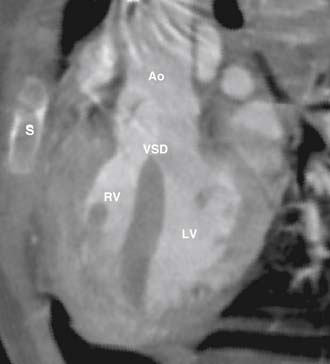
Up to 15% of patients have additional VSDs, which are usually single and muscular, residing in the anterior portion of the septum.15,20 In some cases, an additional VSD exists in the inlet portion of the septum; multiple VSDs are occasionally seen and may be located anywhere in the interventricular septum.
Conduction System
The positions of the sinus and atrioventricular (AV) nodes are normal in patients with TOF, whereas the bundle of His follows the same general course as seen in isolated perimembranous VSDs.14 The bundle of His passes close to the crest of the interventricular septum or slightly to the left of the inferior margin of the VSD. The proximity of the bundle to the crest of the inferior VSD margin is determined by the degree of additional layering in the presence of an extended ventriculo-infundibular fold. When well developed, the ventriculo-infundibular fold and the posterior limb of the trabecula septomarginalis form a continuous muscle border separating the tricuspid anulus from the VSD covering the membranous septum. In these cases, the bundle lies deeply imbedded in muscle, away from the crest of the inferior margin of the VSD, and sutures may be safely placed in this muscle to anchor the patch. When the posterior limb is hypoplastic, the bundle of His comes very close to the crest of the septum, and in these cases, the placement of sutures should be determined by the presence or absence of residual membranous septum. When a generous portion of fibrous membranous septal tissue is present, sutures of the type used for a membranous flap may be safely placed through this fibrous tissue.19,21 When there is a true perimembranous VSD without a membranous flap, sutures must be placed farther down onto the right ventricular aspect of the interventricular septum to avoid the bundle of His. Suture placement in this area contributes to the presence of a right bundle branch block pattern on the postoperative electrocardiogram (ECG), as the bundle of His divides at this point, with the right bundle branch continuing on the rightward aspect of the interventricular septum.22
Coronary Arteries
Significant anomalies of the coronary arteries occur in approximately 5% of patients with TOF.15,23–25 Single coronary arteries arising from the right or left coronary sinuses occur rarely. A single left coronary artery may give rise to the right coronary artery, which may cross the distal RVOT (Fig. 119-9). The most clinically important coronary anomalies occur when large branches from the right coronary artery cross the RVOT. Usually, these are moderate-size conal branches of the right coronary artery, but when they reach the anterior interventricular groove, they may contribute to the coronary arterial supply of the interventricular septum and left ventricle. In rare instances, the anterior descending coronary artery is derived in its entirety from the right coronary artery crossing the RVOT (Fig. 119-10). Rarely, significant anterior descending coronary branches from the right coronary artery cross the RVOT in the infundibular muscle and are not visible on the epicardial surface, making injury to these vessels at the time of TOF repair particularly likely. These anomalies of the coronary arteries have been reported to be more likely to occur when there is anterior and lateral rotation of the aorta in relationship to the pulmonary artery.26 Significant crossing vessels can usually be detected preoperatively by echocardiography, and surgical management can be modified during TOF repair to avoid injury to vessels supplying significant amounts of myocardium.24
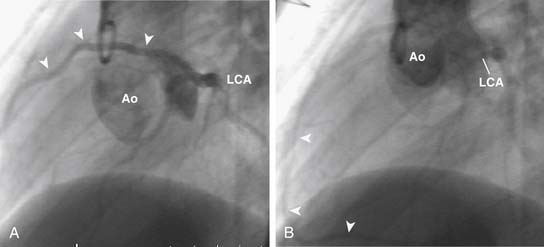
Other Anatomic Features
A right aortic arch is seen in approximately 25% of cases.15 A patent foramen ovale is commonly seen, but a true atrial septal defect (ASD) is probably present only 10% of the time. Major associated lesions are relatively uncommon; multiple VSDs, patent ductus arteriosus, and complete AV septal defects are among the most common.15
CLINICAL FEATURES AND DIAGNOSIS
Laboratory Studies
Polycythemia is seen in most children with chronic cyanosis, and when it is absent, iron deficiency anemia should be suspected. Blood sampling to determine the presence of a deletion on the long arm of chromosome 22 (22qll.2) has become routine during the workup of patients with conotruncal abnormalities. This relatively large deletion is detected by fluorescence in situ hybridization; however, a single gene in the ubiquitin-proteasome pathway involved in apoptosis during embryogenesis may be responsible for the multisystem involvement often observed in these patients.27 Perhaps 10% of patients with TOF and pulmonic stenosis have such a chromosomal deletion, making this the most common syndromic cause of TOF.27,28 There is an even higher incidence of this deletion in patients with TOF and pulmonary atresia, especially in the presence of a right aortic arch and anomalous origin of the left subclavian artery. The presence of this microdeletion has recently been reported to be associated with a more difficult postoperative course in patients with conotruncal defects after surgical repair.29 The chief importance of demonstrating this deletion in a given patient is to provide the opportunity for genetic counseling for future pregnancies and to identify the need for workup of other organ systems that are frequently involved, as summarized by the acronym CATCH-22 (cardiac disease, abnormal faces, thymic hypoplasia, cleft palate, and hypocalcemia associated with a deletion in chromosome 22). Variants of this syndrome have been referred to by a variety of names, including velocardiofacial syndrome, conotruncal anomaly face syndrome, and others. However, the term used to describe its most severe form, the DiGeorge syndrome, is usually reserved for patients with absent thymus, accompanying T-lymphocyte dysfunction, hypocalcemia, and developmental delay.
Echocardiography
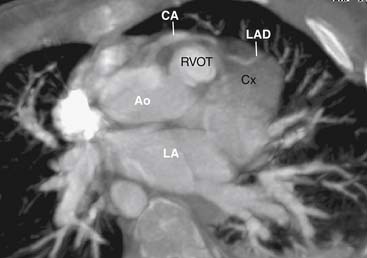
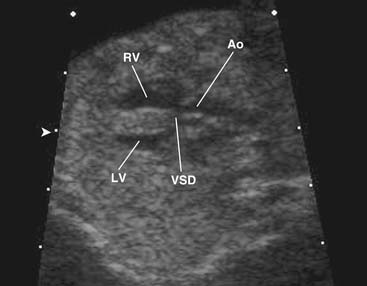
As in most congenital cardiac diagnoses, echocardiography has become the most common and usually definitive diagnostic modality for TOF with pulmonic stenosis. Detailed elucidation of the important anatomic features, including the number and location of VSDs, the nature of the RVOT obstruction, the anatomy of the proximal branch pulmonary arteries, and the coronary artery pattern, can be accomplished with a high degree of accuracy.24,30–32 The ability to acquire definitive anatomic images noninvasively without having to transport the patient is especially important in critically ill neonates, such as those suffering hypercyanotic episodes.
An increasing number of patients are identified in utero with fetal echocardiography (Fig. 119-11).33–41 Prenatal echocardiography can accurately diagnose conotruncal defects such as TOF by the mid to late second trimester.34 Still, many cases remain undetected until after birth, because many pregnancies are not evaluated by prenatal ultrasound and because in those that are evaluated, the diagnosis may be missed with a simple screening scan.40 Prenatally identified cases of TOF may demonstrate poorer outcomes when compared with postnatally diagnosed cases because of a higher proportion with extracardiac defects.38 Generally, prenatal diagnosis of serious congenital heart disease allows delivery to be performed with the appropriate medical and surgical support, which is correlated with improved outcomes.39,41 Perhaps most importantly, prenatal diagnosis of a serious cardiac condition allows the presence of associated conditions such as chromosomal anomalies to be identified, which may have a significant impact on an affected child’s prognosis.
MEDICAL MANAGEMENT
Outpatient Management
A cornerstone of preoperative management is maintaining these infants in a well-hydrated state. Ensuring that these children are on an adequate feeding regimen is extremely important, and prolonged periods without adequate oral intake should be avoided. Every effort should be made to protect affected infants from contacts who have known viral infections. Respiratory viruses such as respiratory syncytial virus can trigger unrelenting hypercyanotic episodes necessitating emergency surgery. Similarly, dehydration accompanying enteroviral diarrheal illnesses can trigger hypercyanotic episodes. Emergency surgery under conditions of an ongoing viral process considerably increases the morbidity and mortality of the procedure. In the absence of underlying pulmonary disease, prolonged oxygen therapy is usually not necessary and, in fact, a new oxygen requirement in a child with previously stable oxygen saturations should suggest that the need for surgery is imminent. Beta-blockers such as propranolol have been used in the outpatient management of children with TOF. The salutary effects of beta-blockers can be attributed to their negative chronotropic effects with increased ventricular filling at slower heart rates. In general, beta-blockers probably provide little in the way of an additional margin of safety in the outpatient management of these children, and their perceived need should prompt a more urgent approach to surgery. However, preoperative use of propranolol appears to have no lasting adverse effects after surgical repair, although additional inotropic support or temporary pacing may be required to overcome mild residual depressive effects on myocardial function or heart rate.42 Diuretics are contraindicated in cyanotic tetralogy, although “pink tets” with relatively unrestricted pulmonary blood flow may require diuretics to treat congestive heart failure.
Balloon Pulmonary Valvuloplasty
Several groups have demonstrated that balloon pulmonary valvuloplasty can be safely performed to palliate infants and children with TOF and pulmonary stenosis.43–46 The majority of cases in these reports had improvement in oxygen saturations, and a delay in surgical intervention was often possible for months after the procedure. Although providing reasonable palliation in selected cases, the role of balloon pulmonary valvuloplasty in TOF remains limited. Balloon pulmonary valvuloplasty may not relieve the multiple levels of obstruction to pulmonary blood flow that are commonly seen in tetralogy patients, especially those who are most symptomatic in infancy. In addition, postprocedure pulmonic insufficiency and catheter-related complications may be significant in these patients. The great majority of infants and children with TOF and severe cyanosis should undergo surgical intervention (as described next), but balloon pulmonary valvuloplasty may occupy a palliative role when immediate surgery is best deferred, as when there is concomitant viral respiratory infection or extreme prematurity.
SURGICAL MANAGEMENT
The Intracardiac Portion of the Repair
Access to the VSD and obstructing muscle bundles in the RVOT is easily achieved by an incision in either the right atrium or the right ventricular infundibulum.15 Viewed through a right atrial incision, the VSD is directly under the septal leaflet of the tricuspid valve with a view underneath the parietal muscle bundles and infundibular septum (Fig. 119-12A). Via a right ventricular incision, the infundibular septum is visualized with the VSD deep to the inferior tip of the infundibular septum and the aortic valve on its posterior surface. When approached through a ventriculotomy, the parietal and septal muscle bundles reside in the lateral recesses above the infundibular septum to the patient’s right and left, respectively. Obstructing parietal muscle bundles are dissected off the ventricular infundibular fold and transected 4 to 5 mm away from the VSD and aortic anulus (toward the right ventricular free wall). A wedge of muscle is removed from this location, particularly in older patients who may have a large amount of hypertrophy of these obstructing muscle bands, although a simple incision of this area may suffice in infants. Obstructing muscle bundle extensions from the infundibular septum along the septal (patient’s left) aspect of the RVOT may also be incised or resected as needed.
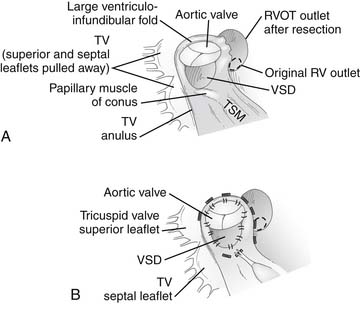
The VSD may then be closed with patch material of either polytetrafluoroethylene (PTFE) or stretch-knitted Dacron after trimming appropriately to shape. Interrupted pledgeted polypropylene (see Fig. 119-12B) or braided nonabsorbable sutures or a running polypropylene suture may be used to sew in the patch. Several critical points of anatomy must be appreciated to ensure complete VSD closure and to minimize the chances for complications. Anteriorly and superiorly, stitches are placed in the fibrous tissue of the aortic anulus to avoid a residual VSD in this region. Inferiorly, if there is a fibrous membranous flap or a prominent muscle bundle from the posterior limb of the trabecula septomarginalis, sutures may be safely placed in these structures. In the absence of these structures, sutures must be placed onto the right ventricular aspect of the interventricular septum to avoid the His bundle. In the absence of a well-developed posterior limb of the trabecula septomarginalis, sutures must be placed in the septal leaflet of the tricuspid valve. If interrupted sutures are used, they should be placed through the tricuspid anulus except in the region of the conducting bundle. This suture is placed a few millimeters from the anulus, to avoid impingement on the AV node by the pledgets. If a continuous suture is used for VSD closure, this portion of the suture line should be performed as a mattress suture to ensure complete closure in this region.
The Pulmonary Artery Portion of the Repair
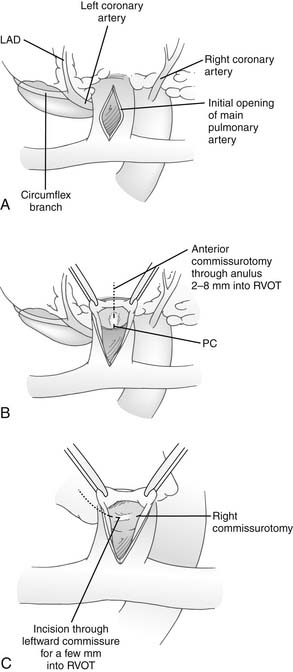
After resection of obstructing muscle bundles in the RVOT, the pulmonary valve is probed using graded dilators. Probing of the pulmonary valve is easily accomplished through either a transatrial approach or a right ventriculotomy. If the pulmonary valve is too small, a longitudinal incision is made in the main pulmonary artery between fine stay sutures (Fig. 119-13). The valve is inspected, full commissurotomies are performed, and the pulmonary valve is probed again to see if it is still inadequate (see later). If it is, a transanular patch will be required, and the valve anulus is incised through the most anterior commissure. If the VSD has been closed through a ventriculotomy, the incision in the pulmonary artery and the incision in the infundibulum are joined through the anterior commissure. If a transatrial approach to the VSD has been used, the incision through the pulmonary valve anulus can often be limited to 2 to 8 mm below the anulus, and further resection of muscle from the RVOT can be performed through the pulmonary artery at that point.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


