Targeted Therapies and Radiation in Lung Cancer
David Raben
Amanda Schwer
Paul Van Houtte
The use of targeted drugs—those that attempt to render specific aspects of the cancer cell-signaling pathway ineffective— have moved forward, although slowly, in the management of advanced non-small cell lung cancer (NSCLC). Erlotinib is currently Food and Drug Administration (FDA) approved for chemorefractory NSCLC.1 Response rates for epidermal growth factor receptor (EGFR) inhibitors are in the range of 9% to 26%,2,3 and recent reports demonstrate that gefitinib has similar activity to docetaxel for second-line treatment in NSCLC.4 These agents fall under the category of drugs that disrupt the EGFR-signaling pathway. Bevacizumab, an intravenously administered humanized monoclonal antibody (MAb), competes with vascular endothelial growth factor (VEGF) and is also active alone or with chemotherapy in advanced NSCLC.5,6 Caution was emphasized when using bevacizumab in patients with central lesions and squamous histology because of increased risk of bleeding. Clinical trials incorporating agents that block VEGF receptor signaling also appear promising when compared to conventional chemotherapeutics.7 So where are we with locally advanced lung cancer and molecularly selective drugs? Have we successfully integrated novel drugs to enhance radiation cytotoxicity? Have we increased toxicity or the therapeutic ratio?
Approaches for stage IIIA to IIIB disease typically include concurrent chemoradiation, which has been found to be superior to radiation alone or sequential chemotherapy and radiation.8,9 Recent evidence suggests that we can indeed push the envelope so to speak and safely deliver much higher doses of radiation (in the range of 74 Gy) when we use extremely conformal techniques.10 When we use conformal intensity-modulated radiation techniques (IMRT) with concurrent chemotherapy, we are reaching median survival rates in the 22-month range.11,12,13 The Radiation Therapy Oncology Group (RTOG) is currently conducting a randomized phase III study (RTOG 0617) that will definitively answer the question of dose escalation by comparing 60 to 74 Gy, both arms incorporating concurrent chemotherapy.
Returning back to targeted agents, we have initiated a host of clinical trials combining EGFR inhibitors or antiangiogenic inhibitors with radiation or chemoradiation. The goals are to see if we can improve survival further without damaging the therapeutic ratio with increased overlapping toxicity. This chapter will review the current and recently completed studies utilizing newer generation molecularly targeted drugs with radiation. Our search for improved outcomes will begin with a review of past and recently published preclinical studies that are driving translational efforts with radiation. Finally, we will take an opportunity to discuss promising agents beyond EGFR and VEGF antagonism, which might also be rationally combined with radiation for lung cancer. A complete review of every study is beyond the scope of this chapter. Rather, we hope to provide some concepts to the reader that may enlighten and/or provoke thought in the area of molecularly targeted agents and radiation for lung cancer.
PRECLINICAL STUDIES WITH MOLECULARLY TARGETED AGENTS AND RADIATION
EGFR Inhibitors and Radiation Why combine EGFR inhibitors with radiation in the first place? We have learned over the past decade or so that there are many reasons to explain the beneficial effects of this combination as seen in preclinical studies. These include cell cycle shifts into phases such as early G1, modest increases in apoptosis, and reduced angiogenesis in vivo. Remember also that ionizing radiation is supposed to damage DNA irrevocably, both through indirect production of free radicals and direct photon interaction with various parts of the DNA matrix, including the base pairs. Cancer cells, however, have a remarkable capacity to repair themselves, and we know that radiation can actually induce this process as well. The hope is that, with the proper amounts of radiation in a clinical setting, through carefully fractionated doses, the cancer cells will be unable to repair all of the damage incurred. Thus, after several cell divisions, repair necessary for mitosis will be incapacitated and the cancer cell will die. EGFR inhibitors also interfere with the repair process, as we will discuss later, and
thus contribute to the enhanced radiosensitization seen with combination therapy.
thus contribute to the enhanced radiosensitization seen with combination therapy.
MAbs with competing activity against EGFR activation have been evaluated extensively in various disease sites. To understand progress to date, we need to review briefly early preclinical studies from over a decade ago starting with anti-EGFR MAbs. We had learned that radiocurability of human tumor xenografts in nude mice expressing EGFR was more difficult than in those without EGFR expression.14 This finding generated a hypothesis that by preventing EGFR activation through prevention of extracellular EGFR dimerization, we might improve radioresponse. Studies performed soon thereafter did find that this was, in fact, the case in tumors expressing EGFR. By combining single or fractionated radiation with concurrent administration of EGFR antibodies, investigators found that they could reduce tumor growth in aerodigestive tract tumors.15,16 These experiments incorporated cetuximab (C225), a human-mouse chimeric MAb, with selective and competitive binding affinity to the extracellular domain of the human EGFR. Investigators began to derive the mechanisms to explain why EGFR interference worked to enhance radiation effects—these included strategic cell cycle blocks at early G1, reduced DNA repair, and modest increases in apoptosis.17 In this same model in vivo, angiogenic abrogation was observed as well. In 2003, further evidence confirming the rationale for blocking EGFR with radiation was evidenced by increased radioresistance observed when an EGFR expression vector was cloned into cancer cells. There was a direct correlation with the level of EGFR expression in the stable clones and radioresistance as measured by clonogenic assays by a factor of 1.28 to 1.6. Cetuximab counteracted this effect by both reducing the levels of EGFR and decreasing phosphorylation of EGFR-related AKT and mitogen-activated protein kinases (MAPK).18
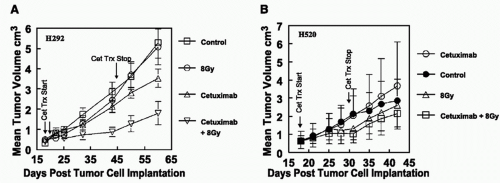 FIGURE 52.1 Mouse models bearing human NSCLC xenografts sensitive (H292; A) and insensitive (H520; B) to EGFR inhibition with cetuximab alone and in combination with single-fraction radiation. Treatment began when tumors had reached approximately 3 cm3. Treatments were cetuximab alone (1 mg/animal twice a week for 2 weeks), ionizing radiation (5 Gy each Monday for 2 weeks), cisplatin (8 mg/kg twice a week for 2 weeks), and combinations of cetuximab plus radiation, cisplatin plus radiation, and cetuximab plus cisplatin plus radiation treatments. Tumor volume ± SE are shown. On day 93, the H292 mean tumor volume was 4.4 cm3 in control animals, 2.2 cm3 in animals treated with irradiation alone (p = 0.049), 1.8 cm3 in animals treated with cisplatin plus radiation (p = 0.024), 1.5 cm3 in animals treated with cetuximab plus radiation (p = 0.012), and 1.2 cm3 in animals treated with cetuximab plus cisplatin and radiation (p = 0.004). The combinations produced a significant reduction in tumor volume compared with controls. Although the combination of all three agents produced the greatest reduction in tumor volume, the differences were not significant compared with cetuximab plus radiation (p = 0.79) or cisplatin plus radiation (p = 0.54). In the H520 xenografts, cetuximab alone and in combination with radiation, cisplatin or the triple combination did not significantly inhibit tumor growth. (From Raben D, Helfrich B, Chan DC, et al. The effects of cetuximab alone and in combination with radiation and/or chemotherapy in lung cancer. Clin Cancer Res 2005;11:795-805.) |
In NSCLC models, there has been only modest exploration of antibodies against EGFR with radiation. Studies at the University of Colorado included evaluation of the EGFR status on NSCLC lines by flow cytometry—little was done at that time with fluorescence in situ hybridization (FISH) to look at EGFR gene amplification—which we have since learned, may be important in predicting response to these agents.19,20,21 Cetuximab monotherapy demonstrated cytostatic effects on some, but not all, NSCLC cell lines with EGFR expression, an effect that appeared to be dose dependent. Cell cycle shifts were noted into the G1 phase, although no effect was seen in cell lines that did not express EGFR. Interestingly, cetuximab actually increased phosphorylated EGFR (pEGFR) in cell lines not stimulated with EGF in cell lines expressing EGFR; however, peak EGF-induced increases in pEGFR were reduced by cetuximab in the cetuximab-sensitive lines. Combination studies in vivo demonstrated cooperative effects between radiation and cetuximab, again only in cell lines sensitive to cetuximab alone, cell lines that did not express EGFR did not demonstrate advantages to combination therapy (Fig. 52.1). Small molecule
inhibitors against EGFR-activated tyrosine kinase inhibitors (EGFR TKIs), including gefitinib and erlotinib, have also shown activity in NSCLC and head and neck squamous cell carcinoma (HNSCC) models. Explanations for their activity include competitive binding with adenosine triphosphate (ATP) to the intracellular tyrosine kinase domain of the EGFR, resulting in reduced receptor phosphorylation and activation.22,23,24 Downstream interference with various pathways ensues, including the Ras-Raf-MAPK and the PI3K/Akt pathways.
inhibitors against EGFR-activated tyrosine kinase inhibitors (EGFR TKIs), including gefitinib and erlotinib, have also shown activity in NSCLC and head and neck squamous cell carcinoma (HNSCC) models. Explanations for their activity include competitive binding with adenosine triphosphate (ATP) to the intracellular tyrosine kinase domain of the EGFR, resulting in reduced receptor phosphorylation and activation.22,23,24 Downstream interference with various pathways ensues, including the Ras-Raf-MAPK and the PI3K/Akt pathways.
In vitro combinations of radiation and erlotinib have resulted in an additive increase in apoptosis in H226 (NSCLC) cells with evidence of increased poly (adenosine diphosphate[ADP]-ribose) polymerase (PARP) cleavage. Erlotinib appeared to subdue radiation-induced pEGFR activation, which might account for rapid repopulation during radiation therapy. Adding to the story was the downregulation of Rad51, an important component of the DNA repair pathway, when erlotinib was added to H226 cells prior to radiation.25 Inhibition of Rad51 has been shown to enhance radiosensitization. Further, erlotinib was also found to augment the effects of suboptimal fractionated radiation in animals bearing H226 flank xenografts.
The EGFR mutation story has received considerable attention over the past several years in regard to small molecule EGFR TKIs.26,27 Specific mutations within the EGFR-binding domain that encode the ATP-binding region, located in exons 18 to 21, have been reported as more prevalent in tumors with adenocarcinoma histology, patients of Asian background, females, and never-smokers.28 Do these same mutations also predict sensitivity or resistance to radiation in lung cancer? Recent evidence suggests that, indeed, mutant EGFR NSCLC cells have dramatically reduced survival rates measured by clonogenic assay compared with wild-type (WT) EGFR NSCLC cells in vitro.29,30 The authors point out that the underlying reasons for this increase in radiosensitivity include delayed DNA repair kinetics. It is theorized that this select group of patients presenting with locally advanced NSCLC could be treated simply with induction EGFR inhibition followed by continued inhibition and radiation.
Adding to the molecular selection story for targeted agents and radiation is the role Kras mutations may play in determining the optimal EGFR-dependent or -independent pathway to block. Lung cancer cells with mutated Kras may drive radioresistance in different ways through EGFR-independent activation of antiapoptotic pathways (PI3K-Akt), thus requiring different targeted agents to effectively enhance radiation cytotoxicity.31 In this regard, blocking AKT signaling appears to enhance radiosensitivity in Kras mutated NSCLC cells primarily through reduced activation of DNA repair pathways, including interference with DNA double-strand break repair.32,33 This certainly provides food for thought in regard to how we might design future clinical trials in locally advanced-stage NSCLC patients with Kras mutations— phase I clinical trials utilizing inhibitors of the AKT pathway with chemoradiation in these patients is the first step.
Angiogenic Inhibitors and Radiation in NSCLC The use of antiangiogenic agents in advanced NSCLC has been validated and accepted by the oncology community. The benefits have been observed primarily in combination studies with chemotherapy34,35 and the gains, although statistically significant, have been modest. As readers may recall, the Eastern Cooperative Oncology Group (ECOG) conducted a randomized phase III study in which 878 patients with recurrent or advanced NSCLC (stage IIIB or IV) were assigned to chemotherapy with paclitaxel and carboplatin alone or with bevacizumab, a humanized antibody against VEGF. This trial was limited only to nonsquamous histologies and patients without brain metastasis caused by bleeding episodes seen in the smaller phase II study. A survival advantage was seen in the bevacizumab arm (12.3 vs. 10.3 months; hazard ratio [HR] for death = 0.79; p = 0.003).35
Is this a valid strategy for radiation therapy for unresectable, locally advanced disease or earlier stage disease patients deemed medically inoperable? The concept of interfering with angiogenesis and the potential benefits and pitfalls with radiation has been explored for many years. The reader is directed toward several reviews that summarize nicely the underlying mechanisms and sequencing issues surrounding the use of antiangiogenics to enhance radiation therapy.36,37,38
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


