The ability to manipulate cell cycle signaling has important clinical implications in lung cancer. For example, modulation of the checkpoints before the completion of DNA repair could enhance cellular sensitivity to DNA damage agents such as radiotherapy, leading to cell death.
This chapter focuses on the cell cycle dysregulation present in lung cancer, the cell cycle effects of radiotherapy, and on the role a cell cycle modulator may play in combination therapy. The knowledge of these concepts might lead to more efficient use of current anticancer therapies and to the development of novel agents.
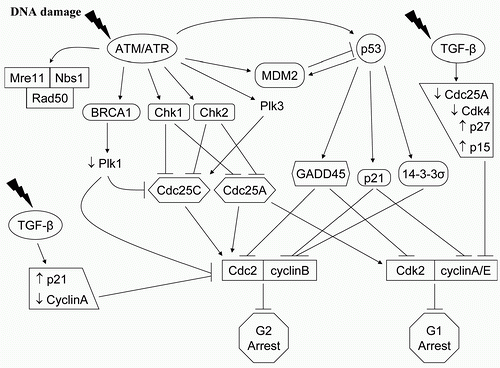
In most solid tumor cells, G 1 arrest is dependent on the activation of the tumor suppressor gene p53 and the downstream CKI p21, which also plays a role in cellular senescence, apoptosis, and DNA repair.
26,
27 It is not clear whether p53
gene and G 1 arrest have an influencing role in radioresistance in solid tumor cells.
28 contrast, G 2 arrest, which can be influenced by p53, may have a radioprotective function by providing cells time to repair DNA damage, and is controlled by the nuclear activities of the cyclin B1-CDC2 complex.
29,
30 Two ATM-dependent G1/S checkpoints can be individualized. First, ATM activation in G1 leads to CHK2 phosphorylation and in turn, to phosphorylation of the phosphatase CDC25A. This increases the proteolytic degradation of CDC25A and prevents activating dephosphorylation of CDK2 and initiation of the G1/S checkpoint.
31,
32 Second, ATM phosphorylates the tumor suppressor p53, either directly or indirectly through CHK2, stabilizing the protein and prolonging its half-life, which function as a transcription factor for the CKI p21. p53 serine 15/20 phosphorylation disrupts the normal binding of the oncoprotein MDM2 to p53, thereby inhibiting its degradation process and prolonging its half-life. This pathway has a role in the maintenance of the G1/S checkpoint.
33Two distinct G2/M checkpoints can also be identified, determined by the kind of DNA damage and by the time sequence.
34 The first checkpoint, ATM dependent, occurs rapidly after radiation-induced damage and is controlled by CHK1-mediated signaling that leads to inhibition of cyclin B1/CDC2 activity. It represents the failure of cells that had been in G 2 at the time of irradiation to progress into mitosis. This G2/M checkpoint, which is dose independent, may be the transducer element linking low-dose hyperradiosensitivity to the subsequent process of resistance.
35 In contrast, the dose-dependent G2/M
accumulation begins to be measurable several hours after irradiation. This mechanism is ATM independent, and represents the accumulation of cells that were in earlier phases of the cell cycle at the time of exposure.
34 G2/M accumulation after exposure to radiation is not affected by the early G2/M checkpoint and is enhanced in cells lacking the radiation-induced S-phase checkpoint, such as those lacking NBS1 or BRCA1 function.
34 It is initiated by the phosphorylation of checkpoint kinases, CHK1 and CHK2, and CDC25A/C, which inactivate the enzymes, blocking activation of CDK1, and causing a G2 arrest. p53 was shown to suppress promoters of cdc2 and cyclin B, which leads to G2/M arrest, and to have a regulation role in the sustain of the G2/M arrest.
36 DNA damage by radiation blocks pRb phosphorylation through p21 to maintain the pRb-regulated cell cycle arrest to complete DNA damage repair. The further identification of cell cycle signaling elements in response to ionizing radiation will have important implication for the development of anticancer targeted strategies.
Cyclins D1 Following a mitogenic signal during the G1 phase, cyclin D1 assembles with cdk4 and a CDK-interacting protein (CIP) (p21)/kinase-interacting protein (KIP) (p27) protein. This complex enters the nucleus and phosphorylates the retinoblastoma tumor suppressor protein Rb1,
37 promoting the release of E2F transcription factor from the Rb1/E2F complex
38 and thus the progression from G1 to S phase. During G1/S transition, glycogen synthase kinase (GSK)-3β enters the nucleus and phosphorylates cyclin D1, allowing its nuclear export by Crm1
39,
40 and degradation by the 26S-proteasome.
40,
41Under certain conditions, the cyclin D1/cdk4 complex can also act as a mediator of programmed cell death.
42,
43 Cyclin D1 can also directly interact with several nuclear receptors and transcription factors,
44,
45,
46,
47,
48 by repressing or inducing them. This transcriptional function links cyclin D1 not only to cell cycle and apoptosis but also to migration, invasion, differentiation, inflammation, and angiogenesis. Cyclin D1 has been identified as a proto-oncogene, PRAD1, and was found to be overexpressed relative to normal tissue in many human cancers.
3,
49,
50 Overexpression of cyclin D1 is associated with dysregulation of cell cycle and is reported in 25% to 60% of non-small cell lung cancer (NSCLC),
51,
52 varying from 30% to 35% in squamous cell carcinoma (SCC) and 36% to 56% in adenocarcinoma (ADC) in the largest series.
53,
54,
55 Cyclin D1 overexpression is less common in neuroendocrine tumors (NE) and, among these, more frequent in atypical carcinoids (20%) and large cell neuroendocrine cancers (LCNE) (9.5%). Cyclin D1 dysregulation is frequently an early event in tumorigenesis, elevated levels being already found in bronchial
56,
57 and alveolar hyperplasia.
58,
59 Cyclin D1 level has also been linked to heavy smoking and said to constitute a marker of tobacco exposure.
52,
60,
61
Cdk Inhibitors
p21. p21 was the first CKI to be discovered. It is a member of the KIP family of proteins that nonspecifically inhibit cyclin-cdk complexes in the nucleus.
62 It can inhibit the cyclin D/cdk4 and cyclin E/cdk2 complexes early in G1, and it can also inhibit the cyclin A/cdk2 complex later, prior to the S-phase/G2-phase transition.
63 p21 transcript is directly activated by p53 and seems to be a fundamental partner for p53. It contributes to p53-induced apoptosis
64 and blocks the G2/M transition through cdc2 inhibition, mediating the pro-apoptotic and cell cycle-arresting effects induced by p53 in response to genotoxic stimuli.
65 Recent studies have
shown potential new roles for p21 and p27, as inhibitors of apoptosis and so, as potential oncogenes. These roles have been related to their relocalization to the cytoplasm,
66,
67,
68,
69,
70 whereas the cell cycle inhibitory activity is dependent on the nuclear localization. Although p21 is infrequently expressed in normal lung,
71 it is overexpressed in 35% to 51% of NSCLC cases, without significant differences between ADC and SCC.
71,
72,
73,
74,
75 p21 expression has been correlated with better differentiation in NSCLC.
71,
73,
74 In two studies adequately controlled for disease stage, p21 expression was associated with improved survival,
72,
75 whereas another study reached slightly different conclusions.
76 In an analysis combining p21 and TGF-β, an improved survival was observed in early stage NSCLC when p21 and TGF-β were in concordance, presenting both high or both low expression.
76 Absence of p21 expression has been correlated to a worse prognosis when associated with p53 positivity
75 and absence of p16 expression.
77p27. p27, another member of the KIP family, blocks the activity of cyclin D1-CDK4/6 and cyclin E-CDK2 and is an important negative regulator of the G1 to S transition.
66,
78 In the normal resting cell, concentration of p27 is high and declines in response to proliferative stimuli. p27 overexpression leads to cell cycle arrest, whereas its loss of expression may result in tumor development and/or progression. As described for p21, some studies have suggested p27 to be an oncogene by inhibiting apoptosis, and that this role is related to its cytoplasmic relocalization.
66,
79 In vivo studies have shown that p27 expression decreases progressively during lung carcinogenesis.
80 Increased p27 proteolytic activity has been associated to low levels of p27.
77,
81 Low p27 expression has been associated with higher stages of lung cancer,
82 as well as poor survival in NSCLC, either by itself
77,
81,
82,
83 and/or when associated to other abnormalities like p53 mutations and/or Rb loss,
84 Ras mutations,
85 high cyclin E levels,
79 high proliferative rates
84,
85 and aneuploidy.
84 A favorable response to chemotherapeutic agents, including drugs targeting EGFR and HER-2, has been correlated to increased p27 expression.
86,
87
p16. p16 is a member of the INK4 family of proteins that inhibits cdk4 and cdk6 activation through a competitive mechanism for cyclin D binding site and specifically during the G1 phase.
88 Loss of p16 has similar effects on G1 progression than overexpression of cyclin D and loss of Rb. The discovery of p16 overexpression in Rb −/− cells, which alters the relationships between cyclin D, cdk4, and cdk6, suggests the existence of a feedback mechanism between p16 and Rb.
89 Decreased p16 is one of the most frequent alterations in lung cancer. Interestingly, loss of p16 is usually found in NSCLC, whereas loss of Rb is found in SCLC. These changes in p16 and Rb seem to be mutually exclusive.
90,
91,
92,
93 The strong inverse correlation is more evident in SCC and SCLC than in ADC.
94,
95
p14. p14 is an alternate transcription product from the INK4/ARF locus shared with p16.
96,
97,
98 By its ability to antagonize the MDM2-mediated ubiquitination and degradation of p53,
99,
100,
101,
102 it induces apoptosis
103,
104 and growth inhibition.
99 p14 is decreased in 50% of NSCLC,
97,
101 where it has an inverse relationship with MDM-2 expression.
101 p14 can be lost secondary to losses at the p16 locus, and also because of the gene promoter methylation.
103,
105
p53 TUMOR SUPPRESSOR. p53, mapped on chromosomic region 17p13, is a tumor suppressor gene that, after sublethal DNA damage, mediates cell cycle arrest in the G1 phase.
106,
107 In this pathway, p53 interacts with several genes. The first one is mdm2: Levels of p53 are kept low by its association with the mdm2 oncogene product, which binds p53 and shuttles it out of the nucleus for proteolytic degradation, establishing an autoregulated feedback circuit. The second gene target is gadd45, that belongs to a gene family implicated in cellular growth arrest.
108,
109 The third target is the gene coding for p21, which develops inhibitory action on multiple cyclin-cdk complexes and also complexes with PCNA, a protein playing a key role in DNA reparation processes.
110 The fourth target is the gene coding for Bax, which promotes apoptotic mechanisms and forms a heterodimer with Bcl-2 gene.
111 As a key regulator of cell growth and cell death, p53 is activated by several kinases that regulate the DNA damage checkpoint following many environmental stimuli, including DNA-damaging agents such as ionizing radiation, and is crucial in preventing the propagation of mutations in normal cells.
67 Activated p53 induces cell cycle arrest (p21
Cip1/Waf1) to allow cells to repair the damage or apoptosis if the damage is too severe and/or irreparable.
112 During carcinogenesis, p53 is frequently inactivated by multiple mechanisms. The most common mechanism is mutation at the p53 gene, which occurs in more than 50% of all human cancers. In response to DNA damage, some p53 mutants show less capacity to bind and initiate transcription from their target genes, such as p21, Mdm2, Bax, and cyclin G, and so, some of the p53-mediated effects are blunted,
113 resulting in insensitivity to growth-inhibitory signals as well as evasion of apoptosis. Mouse models have confirmed cooperation between mutated p53 and mutative active K- ras in NSCLC development
114 and between p53 and Rb in SCLC development.
115
p53 mutation is one of the most common genetic alterations in lung cancer, with about 70% of SCLC and 50% of NSCLC presenting mutations in one allele of p53, often accompanied with loss of the wild-type allele.
116 In NSCLC, p53 mutations are more frequently found in SCC than ADC.
117 The p53 mutational spectra of lung cancer are different from those of other cancers with an excess of G:C to T:A observed,
118 these ones especially linked to exposure to tobacco carcinogens such as benzopyrene.
119 Several studies investigated the association of p53 abnormalities with prognosis in lung cancer, with discordant results. Some of them showed an association of p53 overexpression (mutants form have a longer half-life and lead to the detection of high levels of protein by IHC) with shorter survival,
120,
121,
122 others failed to find such an association.
123,
124 Concurrent p53 and p16 abnormalities have been associated to a worse prognosis.
125,
126 Many reports have linked the presence of p53 mutations to resistance to DNA-damaging chemotherapeutics agents,
127 and this is not surprising as we know that most chemotherapeutics agents act by stimulating apoptosis.
Cell Cycle as Target for Combined Treatment with Radiotherapy Cellular radiosensitivity varies along the different phases of cell cycle. The majority of cells surviving after a first dose of radiation were most likely in a less sensitive (G1 phase) or in a resistant phase of the cell cycle (late S phase). Those cells will then progress into a more sensitive cell cycle phase, that is at or close to mitosis, which represents a more ideal time for delivering radiation therapy. Several cell cycle regulatory proteins are potential molecular targets for cancer therapy, and agents can be combined to radiation to enhance its effects on lung cancer. Efforts have been made to sensitize cancer cells to the cytotoxicity of DNA damage by anticancer agents as early as four decades ago with compounds such as caffeine, which resulted in abrogation of the G2 cell cycle checkpoint. Many malignant cells, including lung cancer, have defective G1 checkpoint mechanisms and depend far more on G2 checkpoint than normal cells. More recently, several potent agents have been studied in preclinical and clinical trials. Among them is flavopiridol, the first CKI to enter clinical trials as a potential anticancer agent that exerts both cytostatic and cytotoxic actions on cancer cells. Another well-known drug is UCN-01, a potent prototypical chk1 inhibitor, which function as a CKI at high concentrations. Such compounds seek to force cells to enter mitosis without allowing adequate time for DNA repair, increasing the likelihood that cell death will occur by accumulation of DNA lesions. Treatment strategies consisting in abrogation of G2 checkpoints can also be achieved with the use of microtubule-targeting compounds, such as taxanes and epothilones to stall cells in G2/M (by preventing mitosis, thereby trapping cells in the phase of the cell cycle where ionizing radiation have the greatest effects). Microtubule-stabilizing agents will be discussed as mitotic targets in combination with ionizing radiation. Finally, Aurora kinases, part of the spindle checkpoint, are recent agents that can also be used in combined anticancer therapy (
Table 14.2).
Cyclin-Dependent Kinases (CDK) Modulators Cell cycle regulatory proteins such as CDKs are potential molecular targets for radiation therapy.
128,
129,
130 The rationale for targeting the cell cycle and the CDKs in lung cancer therapy has been based on the frequency of their perturbations in lung tumors (overexpression of cyclins, and/or absent or diminished levels of CKI).
54,
55,
131,
132 For example, overexpression of cyclin D1 and loss of p16 gene expression has been associated with the development of lung cancer (see previous discussion). These defects in tumor cells lead to uncontrolled proliferation as a result of the loss of checkpoint integrity. In addition, cell cycle arrest by CKI has been shown to induce apoptosis.
1,
133,
134,
135
FLAVOPIRIDOL. Flavopiridol is a semisynthetic flavone that directly competes with the ATP substrate and reversibly inhibits kinase activity of multiple classes of CDKs, including cdk1, cdk2, cdk4, cdk6, cdk7, and cdk9 at submicromolar concentrations (IC50 values of 100 to 400).
136 This pancyclin inhibitor causes arrest at both G1 and G2/M phases of the cell cycle by several mechanisms: direct inhibition of cdks 1, 2, and 4, and indirect inhibition by downregulating cyclin H-cdk7,
137,
138,
139,
140 as well as tumor growth arrest in most solid tumors and xenografts.
140,
141,
142,
143,
144 Flavopiridol also promotes a decrease in the level of cyclin D1,
135 which is commonly overexpressed in many cancers including lung cancer where it has been described as a poor prognosis marker.
51,
52,
53,
54,
55,
131,
145,
146,
147,
148,
149 However, at considerably higher concentrations than necessary to inhibit CDKs, flavopiridol inhibits the activity of several other protein kinases
150,
151 including signal transducing kinases protein kinase A (PKA), PKC, and Erk-1, the receptor tyrosine kinase EGFR, and receptor-associated protein kinases such as c-Src.
152 In addition, although described as cytostatic, flavopiridol has been shown to be also cytotoxic to many lung cancer cell lines
143,
144,
153,
154 by induction of apoptosis except in the A549 lung carcinoma cell line.
154 Normally, DNA-damaging agents induce p53, which in turn transcriptionally induces p21 and Mdm-2, the later binds p53 and targets it for degradation.
155 Although upregulating p53, flavopiridol was shown to inhibit transcription of p21 and Mdm-2 and to inhibit cell proliferation in A549 lung cancer cells.
153
Despite promising preclinical data, flavopiridol has not demonstrated in trial significant clinical activity as a single agent in patients with metastatic lung cancer.
129 The unexpected and significant toxicity of this agent given as single cancer therapy in all these clinical trials have so far discouraged its use in monotherapy.
Another approach for the use of flavopiridol in anticancer therapy is to take advantage of its potential to augment cytotoxic actions of chemotherapeutic agents and radiation. This strategy of combining flavopiridol with chemotherapy has been investigated in several studies showing promising results. Flavopiridol enhanced the cytotoxic effects of many chemotherapeutic agents in vitro (12, 13, 14, 15, 16, and 17) as well as in vivo (8, 13, and 15), in a sequence-dependent manner. Similar results were observed in the combined approach with ionizing radiation both in vitro,
156,
157 and in vivo
130 in various human cancer types.
156,
158,
159,
160 Flavopiridol sensitized human cancers to radiation in a dose-dependent manner, by cell cycle redistribution, by inhibiting cellular repair from radiation damage, and possibly by effects on angiogenic factors. In addition, studies also demonstrated the effects of flavopiridol in enhancing apoptosis and tumor regression.
160,
161,
162,
163 The therapeutic ratio of radiotherapy in the in vivo tumor models was increased by flavopiridol.
130,
156 More specifically, the radiosensitizing action of flavopiridol was recently determined in zebrafish embryos using cyclin D1 (CCND1) downregulation by antisense hydroxylprolyl-phosphono peptide nucleic acid oligomers compared to control.
164 This study demonstrated that the specific sensitizing effects of flavopiridol in response to radiation were in part caused by the inhibition of cyclin D1, one of its primary pharmacologic targets.
158 Another study analysed the sequence-dependent effects of flavopiridol when combined to radiation and showed that the maximum radiopotentiation and apoptosis were observed when the lung cancer cells were treated with the sequence of docetaxel, then radiation, and finally flavopiridol both in vitro and in vivo. Therefore, the combined, sequence-dependent strategy radiation/flavopiridol has the potential to enhance outcome in many types of cancer and needs to be further investigated in a well-designed clinical trial.