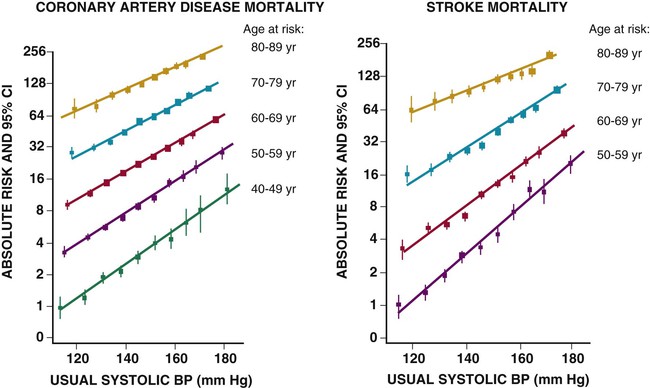
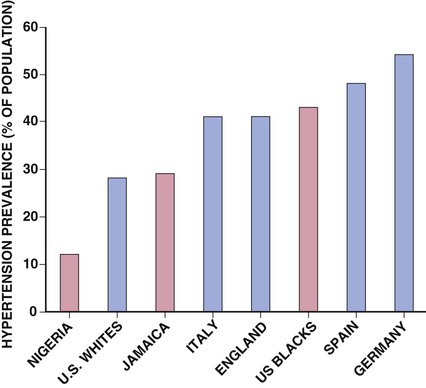
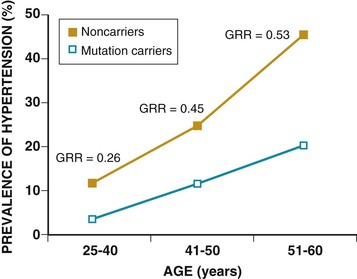
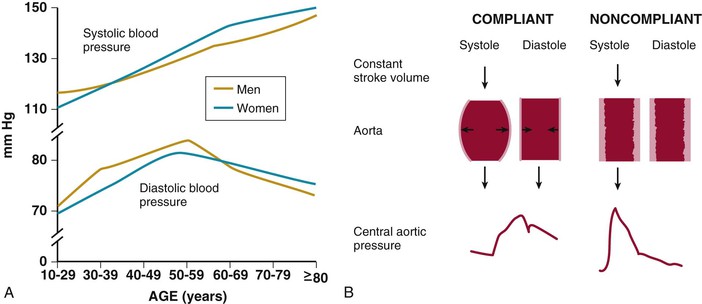
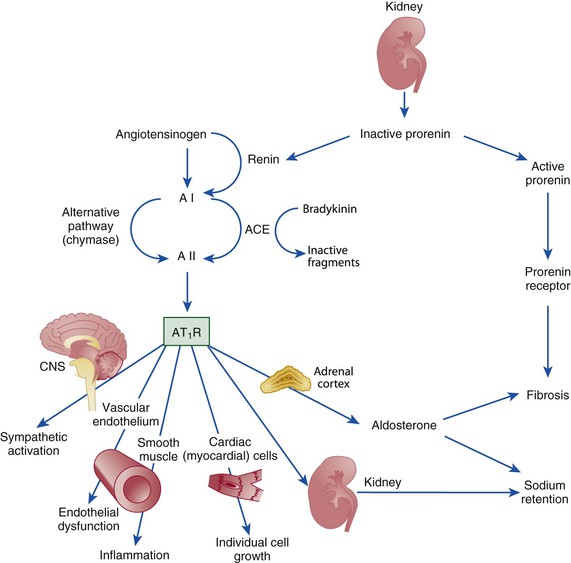
Ronald G. Victor Affecting 75 million people in the United States and 1 billion worldwide, hypertension remains the most common, readily identifiable, and reversible risk factor for myocardial infarction (MI), stroke, heart failure, atrial fibrillation, aortic dissection, and peripheral arterial disease (see also Chapters 1 and 42). The global burden of hypertension is rising owing to escalating obesity and population aging, and the condition is projected to affect 1.5 billion persons—one third of the world’s population—by 2025. The prevalence of hypertension is increasing rapidly in developing countries, where poor hypertension treatment and control contribute to the growing epidemic of cardiovascular disease.1 High blood pressure currently causes two thirds of all strokes and half of all cases of ischemic heart disease worldwide.2 Half of this disease burden occurs in people with hypertension (i.e., blood pressure ≥ 140/90 mm Hg); the other half occurs in people with lesser degrees of high blood pressure (prehypertension). Thus high blood pressure remains the leading cause of death worldwide and one of the world’s great public health problems (see Chapter 1). The asymptomatic nature of this condition delays diagnosis. Effective treatment requires continuity of care by a knowledgeable clinician and frequent medical checkups, which are less common in men and in members of low-income minority groups.3 Most patients diagnosed with hypertension do not manifest a single disease-causing mechanism. Treatment therefore remains empirical, often requiring three or more pharmacologic agents with complementary mechanisms of action along with lipid-lowering drugs, antiplatelet drugs, and drugs for concomitant medical conditions such as diabetes. Pill burden, prescription drug costs, medication side effects, and insufficient time for patient education contribute to medication nonadherence. Physicians often undertreat hypertension (see Chapter 44).4 For all of these reasons, blood pressure remains elevated—140/90 mm Hg or higher—in more than half of affected persons in the United States5 and other developed countries. Even among patients whose hypertension control meets current standards, fewer than one in three is protected from subsequent stroke, MI, or heart failure. The resultant annual cost to the U.S. health care system exceeds $73 billion, yielding a global health care cost of some $3.6 trillion.6,7 This chapter and Chapter 44 review the scientific basis for current recommendations for the diagnosis, evaluation, and treatment of hypertension and present emerging concepts from clinical and basic research that affect clinical decision making. Hypertension is defined as a usual office blood pressure of 140/90 mm Hg or higher.8 Yet epidemiologic data show continuous positive relationships between the risk of coronary artery disease (CAD) and stroke deaths with systolic or diastolic blood pressure down to values as low as 115/75 mm Hg9 (Fig. 43-1). The artificial dichotomy between “hypertension” and “normotension” may delay medical treatment until irreversible compromise of vascular health by elevated blood pressure values previously considered normal. On the other hand, the current body of evidence from randomized controlled trials does not permit experts to achieve consensus on whether to recommend blood pressure–lowering medications for high-risk patients with blood pressure in the “prehypertensive” range of 120 to 139/80 to 89 mm Hg. In the United States and other developed countries, the prevalence of hypertension increases with age—rising exponentially after 30 years of age (see Chapter 1). Before 50 years of age, women have a somewhat lower prevalence of hypertension than men. After menopause, the prevalence of hypertension increases rapidly in women and surpasses that in men. Eventually, by 75 years of age—below the average life span of U.S. men and women—almost 90% will have hypertension. More than 40% of non-Hispanic black adults in the United States have hypertension, compared with 25% of non-Hispanic white and Hispanic adults. Black Americans also have earlier onset and more severe hypertension and suffer greater target organ damage, leading to greater premature disability and death. Hypertension and its complications are even more prevalent in many predominantly white European countries than in black Americans but far less prevalent among black Africans10 (Fig. 43-2). Hypertension prevalence does not vary between black and non-black Hispanic adults in Cuba. Although genetic factors may explain the disproportionate burden of hypertension in black Americans, these international data underscore the importance of environment. From 90% to 95% of hypertensive patients have no apparent single reversible cause of elevated blood pressure, hence the term primary hypertension. The remaining 5% to 10%—cases designated as secondary or identifiable hypertension—demonstrate a more discrete mechanism. In most patients with primary hypertension, readily identifiable behaviors contribute to the elevated blood pressure. The nicotine in cigarette smoke transiently raises blood pressure by 10 to 20 mm Hg, thereby elevating the average daytime blood pressure in habitual smokers. Moderate alcohol drinkers (one or two drinks per day) generally have less hypertension than teetotalers, but the risk for development of hypertension increases in heavy drinkers (three or more drinks per day). Hypertension is rare in Asian men who abstain from alcohol to avoid the nausea and flushing reaction associated with their loss-of-function mutation in the alcohol dehydrogenase gene (ALDH2).11,12 Caffeine consumption typically causes only a small transient rise in blood pressure, which in some persons habituates after the first cup of coffee. The risk for development of hypertension does not vary with coffee consumption but increases steeply when caffeine is consumed in diet sodas; thus coffee may contain protective antioxidant polyphenols not present in sodas. Physical inactivity also increases the risk for developing hypertension. Lifetime dietary habits clearly influence the risk for developing hypertension (see Chapters 44 and 46). Diets low in fresh fruit may increase risk, but excessive consumption of calories and sodium are the two most important behavioral determinants of hypertension. Across various populations, hypertension prevalence increases linearly with average body mass index. Currently, up to 50% of all cases of hypertension may result from obesity. The risk for developing hypertension increases with dietary sodium intake and decreases with dietary potassium intake.13 Individual variability in blood pressure responses to dietary sodium loading and sodium restriction indicates an important genetic underpinning. Concordance of blood pressure is higher in families than in unrelated persons, higher for monozygotic than for dizygotic twins, and higher among biologic than among adoptive siblings living in the same household. As much as 70% of the familial aggregation of blood pressure may result from shared genes rather than to shared environment (see Chapter 8). The complex regulation of blood pressure has thwarted the genetic dissection of primary human hypertension. Although mutations in 20 salt-handling genes cause ultrarare monogenic forms of severe early-onset hypotension (salt-wasting syndromes) and hypertension (all inherited as mendelian traits), applicability to common primary hypertension has proved elusive. Data from the Framingham Heart Study indicate that 1% to 2% of the general adult population has gene mutations underlying the pediatric salt-wasting syndromes (Bartter and Gitelman syndromes) that may confer resistance against primary hypertension14 (Fig. 43-3). Worldwide research consortia of genome-wide association studies confirmed eight loci for blood pressure, but the individual effect size for each is so small that together these loci explain less than 1% of blood pressure variance. The large gap between estimated and observed variance—termed “missing heritability”—could be due in part to “epigenetics,” which refers to the inheritance of gene expression patterns not strictly dependent on differences in DNA sequence.15 Primary hypertension falls into three distinctly different hemodynamic subtypes that vary sharply by age. Typically associated with hypertension in the elderly (see later), isolated systolic hypertension (ISH) also is the main type in young adults (typically 17 to 25 years of age). The key hemodynamic abnormalities are increased cardiac output and a stiff aorta, both presumably reflecting an overactive sympathetic nervous system. The prevalence may reach as high as 25% in young men, but the condition affects only 2% of young women. Several recent studies show that young persons with ISH have elevated central as well as brachial systolic blood pressures, indicating significantly increased hemodynamic burden.16 Thus ISH in youth may predispose to diastolic hypertension in middle age. Hypertension diagnosed in middle age (typically, 30 to 50 years of age) usually has the elevated diastolic pressure pattern, with normal systolic pressure (isolated diastolic hypertension) or elevated systolic pressure (combined systolic-diastolic hypertension). This pattern constitutes classic “essential hypertension.” Isolated diastolic hypertension is more common in men and often associates with middle-age weight gain. Without treatment, isolated diastolic hypertension often progresses to combined systolic-diastolic hypertension. The fundamental hemodynamic fault is an elevated systemic vascular resistance coupled with an inappropriately normal cardiac output. Vasoconstriction at the level of the resistance arterioles results from increased neurohormonal drive and an autoregulatory reaction of vascular smooth muscle to an expanded plasma volume, the latter because of impairment in the kidneys’ ability to excrete sodium. After the age of 55 years, ISH (systolic blood pressure > 140 mm Hg and diastolic blood pressure < 90 mm Hg) predominates. In developed countries, systolic pressure rises steadily with age; by contrast, diastolic pressure rises until approximately 55 years of age and then falls progressively thereafter (Fig. 43-4). The resultant widening of pulse pressure indicates stiffening of the central aorta and a more rapid return of reflected pulse waves from the periphery, augmenting systolic aortic pressure (see Fig. 43-4, also see Figs. e43-1, e43-2, and e43-3 ISH may represent an exaggeration of this age-dependent stiffening process, although systolic blood pressure and pulse pressure do not rise with age in the absence of urbanization (e.g., cloistered nuns). ISH is more common in women and associates prominently with heart failure with preserved systolic function, a syndrome also more prevalent in women (see Chapters 27, 76, and 77). Most cases of ISH arise de novo after 55 years of age and do not represent “burned-out” middle-age diastolic hypertension; more than 80% of patients with isolated diastolic hypertension, however, will develop ISH in the next decade of life.16 Compared with young or middle-aged adults with optimal blood pressure, those with pressures in the high-normal range (prehypertension) are more likely to develop ISH after the age of 55 years. A multitude of neurohormonal, renal, and vascular mechanisms interact to various degrees in contributing to these different hemodynamic forms of hypertension. Two invasive approaches to treat hypertension—surgical implantation of a carotid baroreceptor pacemaker and catheter-induced renal nerve ablation (see Chapter 60)—have rekindled great interest in the neural mechanisms of clinical hypertension.18,19 Figure 43-5 shows the major central and reflex mechanisms thought to drive sympathetic overactivity in human hypertension. These include, among others, resetting of the baroreceptors and activation of renal sensory nerves termed renal afferents. Figure 43-5 also shows the specific mechanisms that are targeted by the device-based therapies. Neither device is yet approved by the U.S. Food and Drug Administration (FDA). The Rheos system (CVRx, Inc., Minneapolis) is a surgically implanted carotid baroreceptor pacemaker.20 With the patient under general anesthesia, electrode wires are implanted around the carotid sinus nerves in the neck and connected to a pacemaker generator placed in a subcutaneous pocket in the chest. Electrical stimulation of the carotid sinus nerves sends afferent neural signals that the brainstem interprets as a rise in blood pressure, evoking a reflex reduction in blood pressure. The efferent arm of this reflex arc involves decreased efferent sympathetic nerve activity to the heart, which slows heart rate; to the peripheral circulation, which lowers systemic vascular resistance; and to the kidney, which reduces renin release and increases renal sodium excretion. Activation of the Rheos device acutely decreases sympathetic nerve activity, blood pressure, and heart rate and may avert acute hypertensive crisis.21 Although the carotid sinus and aortic arch baroreceptors buffer acute increases in blood pressure, data regarding the durability of the antihypertensive action of continual carotid baroreceptor stimulation are lacking. This question was addressed by the Rheos Pivotal Trial, a randomized, double-blind, placebo-controlled study of carotid baroreceptor pacing in patients with drug-resistant hypertension.20 In the Rheos trial, 265 patients with resistant hypertension and baseline blood pressure averaging 169/101 mm Hg (despite treatment of most patients with five or more blood pressure medications) underwent implantation of the Rheos device and subsequently random assignment (2:1) 1 month after implantation to immediate initiation of bilateral carotid baroreceptor pacing (group A) or delayed initiation until the 6-month visit (group B); all patients received open-label baroreceptor pacing for another 6 months. The results were largely negative, but mixed. There were no group differences at 6 or 12 months in the coprimary endpoints of percentage of subjects in whom systolic blood pressure decreased by at least 10 mm Hg (54% for group A and 46% for group B; P = NS); and 9% of patients developed transient or permanent facial nerve injury. Yet a post-hoc analysis showed that 42% of group A patients and 24% of group B patients achieved systolic blood pressure control (systolic blood pressure ≤ 140 mm Hg) at 6 months (P = 0.005), with just over 50% of both groups achieving systolic blood pressure control at 12 months (at which point group B had received baroreceptor pacing for 6 months). The reduction in blood pressure associated with a small initial decrease in estimated glomerular filtration rate (eGFR).22 More research should determine if efficacy and safety can be improved by additional technical refinements or if offsetting responses from aortic baroreceptors, which are not paced, inherently limit this approach. A second-generation minimally invasive unilateral carotid nerve pacing system (Barostim neo) has yielded encouraging preliminary results for safety and efficacy.23 Rat studies have identified a major role for the renal sympathetic nerves in the development of hypertension, but the importance of the renal nerves in causing human hypertension previously has not been studied directly. Renal sympathetic nerves cause renal vasoconstriction and vascular hypertrophy via alpha-1 receptors, stimulate renin release via beta-1 receptors, and enhance renal sodium and water reabsorption via alpha-1 receptors (Fig. 43-6). Thus catheter-based renal denervation is an exciting novel approach to treat patients with drug-resistant hypertension, as described and applied in the Symplicity HTN-1 and HTN-2 trials.24 Radiofrequency current delivered by way of an intraluminal catheter destroys the renal nerves, located on the adventitial surface of the renal arteries. With the patient under conscious sedation, the Symplicity catheter is advanced into each renal artery, and four to six discrete low-power radiofrequency treatments are applied along the length of each artery. In the unblinded Symplicity HTN-2 trial (Renal Denervation in Patients with Uncontrolled Hypertension), a total of 106 non-U.S. patients with drug-resistant hypertension and baseline blood pressure of 178/97 mm Hg despite treatment with an average of five or more blood pressure medications randomly were assigned to undergo renal denervation while continuing previous drug therapy or to continue previous drug therapy alone. Patients who met initial screening criteria were excluded if systolic blood pressure fell below 160 mm Hg on a second screening or if they had unfavorable renal anatomy. The primary endpoint was the change from baseline in seated office-based measurement of systolic blood pressure at 6 months. During the intervention, a catheter was inserted into the renal arteries with the patient under conscious sedation, and four to six discrete low-power radiofrequency treatments were applied along the intraluminal length of both renal arteries; the goal was to cause thermal destruction of the renal nerves, which are located on the adventitial (outer) surface of the renal arteries. Office-based blood pressure fell dramatically by 32/12 mm Hg in the active treatment group, versus no change in the control group. The 24-hour ambulatory blood pressure, measured in less than half of patients, fell less dramatically: by 11/7 mm Hg in the active treatment group, versus no change in the control group. No major adverse events occurred. Subsequently, smaller studies have suggested multiple ancillary benefits of renal denervation, including improvement in glycemic control, sleep apnea,25,26 and quality of life27; regression of left ventricular mass28; reduction in central aortic stiffness29,30; and adjunctive treatment for atrial fibrillation.30 These diverse benefits presumably derive not from destruction of efferent renal sympathetic nerve fibers but rather from destruction of renal afferent (sensory) nerves—thereby causing a global reduction in sympathetic outflow to multiple tissues and vascular beds19 (Fig. 43-7). The new 3-year follow-up data from the open-label uncontrolled Symplicity HTN-1 trial in 88 patients show impressive sustained reductions in office blood pressure averaging −36/−14 mm Hg, but ambulatory blood pressure was not assessed.31 Renal denervation did not prevent a decline in eGFR, but without a control group, we do not know if this decline is less than or greater than that caused by hypertension alone without renal denervation. Based on the outcomes of the randomized but open-label Symplicity HTN-2 trial, renal denervation already has been approved for clinical use in Europe, Australia, and Asia. The randomized and blinded Symplicity HTN-3 trial, however, did not show a significant reduction in office or ambulatory blood pressure 6 months post procedure compared to a sham intervention.31b Thus validation of the efficacy of this procedure will require further work. Renal denervation studies already have proven an important sympathetic neural contribution to severe drug-resistant human hypertension. Previously, the sympathetic nervous system was implicated mainly in the initiation of hypertension, but not in its maintenance. These studies also suggest a greatly expanded role of the renal afferents, formerly thought to contribute mainly in renal parenchymal hypertension and cyclosporine-induced hypertension.32,33 In young adults, primary hypertension consistently associates with increased heart rate and cardiac output, plasma and urinary norepinephrine levels, regional norepinephrine spillover, peripheral postganglionic sympathetic nerve firing (determined by microelectrode recordings), and alpha-adrenergic receptor–mediated vasoconstrictor tone in the peripheral circulation.18 Sympathetic overactivity occurs in early primary hypertension and in several other forms of established human hypertension, including hypertension associated with obesity, sleep apnea, early type 2 diabetes mellitus and prediabetes, chronic kidney disease (CKD), heart failure, and immunosuppressive therapy with calcineurin inhibitors such as cyclosporine. In these conditions, central sympathetic outflow can result from deactivation of inhibitory neural inputs (e.g., baroreceptors), activation of excitatory neural inputs (e.g., carotid body chemoreceptors, renal afferents), or circulating angiotensin II (A II), which activates pools of excitatory brainstem neurons without a blood-brain barrier (see Fig. 43-5). In hypertension, the baroreceptors reset to defend a higher level of blood pressure. Baroreflex control of sinus node function is abnormal even in mild hypertension, but baroreflex control of systemic vascular resistance and blood pressure is well preserved until diastolic function is impaired.34 Complete baroreflex failure (see Chapter 89) causes labile hypertension, most often seen in throat cancer survivors as a late complication of radiation therapy, which causes a gradual destruction of the baroreceptor nerves. Partial baroreceptor dysfunction is common in elderly hypertensive patients and typically manifests with a triad of orthostatic hypotension, supine hypertension, and symptomatic postprandial hypotension—the last initiated by splanchnic pooling after carbohydrate-rich meals. Neural mechanisms of obesity-related hypertension deserve special mention. With weight gain, reflex sympathetic activation may be an important compensation to burn fat, but at the expense of sympathetic overactivity in target tissues (i.e., vascular smooth muscle and kidney) that produces hypertension. Hypertensive patients with the metabolic syndrome, with or without new-onset type 2 diabetes, have near-maximal rates of sympathetic firing. Although the sympathetic activation associates with insulin resistance, the precise stimulus to sympathetic outflow is unknown; candidates include leptin, other adipokines, and A II. Why weight loss improves hypertension much less than diabetes remains unknown.35 Patients with obstructive sleep apnea can have markedly elevated plasma and urine catecholamine levels, mimicking those seen in patients with pheochromocytoma (see Chapter 81). With repeated arterial desaturation during apneas, activation of carotid body chemoreceptors causes dramatic pressor episodes throughout the night and resets the chemoreceptor reflex; daytime normoxia is misinterpreted as hypoxia, producing sustained reflex sympathetic activation and hypertension even during waking hours (see Chapter 75). Obstructive sleep apnea also accelerates the risk of several hypertensive complications (e.g., stroke, atrial fibrillation, and cardiovascular death) beyond that explained by blood pressure elevation alone.36 The typical U.S. diet is high in NaCl, with most dietary salt coming from processed food (see also Chapter 46). Although men consume an estimated 10.7 g of NaCl daily, and women 7.3 g, the Department of Agriculture and the Department of Health and Human Services recommend a daily intake of less than 5.8 g of NaCl (2300 mg of sodium) for the general population, and 3.7 g for persons with hypertension or prehypertension. If the food industry agreed to a palatable reduction in the salt content of processed food, reducing dietary salt by 3 g per day probably would reduce the annual number of new cases of coronary heart disease by 60,000 to 120,000, new cases of stroke by 32,000 to 66,000, and new cases of MI by 54,000 to 99,000 and would reduce the annual number of deaths from any cause by 44,000 to 92,000. All segments of the population would benefit, with blacks benefiting proportionately more, women benefiting particularly from stroke reduction, older adults from reductions in coronary heart disease events, and younger adults from lower mortality rates.37 In many forms of experimental and human hypertension, the fundamental abnormality is an acquired or inherited defect in the kidneys’ ability to excrete the excessive sodium load imposed by a modern diet high in salt. Because humans evolved in a low-sodium/high-potassium environment, the human kidney handles poorly exposure to high sodium and low potassium. Renal sodium retention expands the plasma volume, increasing cardiac output and triggering autoregulatory responses that increase systemic vascular resistance. Salt retention also augments the smooth muscle contraction produced by endogenous vasoconstrictors. Beyond raising blood pressure, a high-salt diet also accelerates hypertensive target organ damage. In normotensive persons, blood pressure elevation invokes an immediate increase in renal sodium excretion to shrink plasma volume and to return blood pressure to normal. In almost all forms of hypertension, the pressure-natriuresis curve is shifted to the right, and in salt-sensitive hypertension, the slope is reduced. Resetting of the pressure-natriuresis curve prevents the return of blood pressure to normal, so that fluid balance is maintained but at the expense of high blood pressure. It also leads to nocturia, one of the most common and bothersome symptoms in patients with uncontrolled hypertension. Hypertensive persons excrete the same amount of a given dietary sodium load as normotensive persons do, but at a higher blood pressure, and require many more hours to excrete the sodium load and to achieve sodium balance. Renal inflammation is both the cause and the consequence of renal medullary ischemia, the hallmark of both initiation and progression of salt-dependent hypertension in rodents. Consequent to fetal undernutrition, low birth weight with reduced nephrogenesis increases the risk for development of adult salt-dependent hypertension. This association does not depend on shared genes, shared postnatal environment, or risk factors for adult hypertension. Hypertensive adults have fewer glomeruli per kidney but very few obsolescent glomeruli, suggesting that nephron dropout with decreased total filtration surface area is the cause and not the consequence of the hypertension. When low-birth-weight children are exposed to a fast-food diet, they are susceptible to rapid weight gain, leading to adolescent obesity and hypertension. Animal and human studies have implicated an important genetic contribution to salt-sensitive hypertension. Rats with inbred defects in the kidneys’ ability to excrete sodium remain relatively normotensive on a sodium-restricted diet, but become severely hypertensive when fed a high-sodium diet—a model of salt-sensitive hypertension that can be cured by interstrain renal transplantation. A similar gene-environment interaction may explain why persons of sub-Saharan African ancestry remain normotensive on a sodium-restricted diet but are predisposed to the development of hypertension when they are exposed to a high-sodium diet. Ancestral gene analysis has not ascertained the molecular basis for salt-dependent human hypertension but has identified a common genetic predisposition in African-origin populations to all nondiabetic forms of CKD, including focal glomerulosclerosis, acquired immunodeficiency syndrome (AIDS), and hypertensive nephropathy. Sequence variations in the APOL1 gene are strongly associated with African ancestry and confer a twofold to fourfold increased risk of end-stage renal disease, independent of blood pressure.38 As the kidneys fail, blood pressure becomes increasingly salt-dependent. Alterations in the structure and function of small and large arteries are pivotal in the pathogenesis and progression of hypertension. The endothelial lining of blood vessels is critical to vascular health and constitutes a major defense against hypertension. Dysfunctional endothelium displays impaired release of endothelium-derived relaxing factors (e.g., nitric oxide, endothelium-derived hyperpolarizing factor) and enhanced release of endothelium-derived constricting, proinflammatory, prothrombotic, and growth factors39 (Fig. 43-8). The endothelium of all blood vessels expresses the enzyme nitric oxide synthase, which can be activated by bradykinin, acetylcholine, or cyclic laminar shear stress. Nitric oxide synthase generates nitric oxide, a volatile gas that diffuses to the adjacent vascular smooth muscle and activates a series of G kinases that culminate in vasodilation (see Fig. 43-8). In humans, endothelium-dependent vasodilation can be assessed by measuring increases in the large artery (forearm or coronary) diameter after intra-arterial infusion of acetylcholine or release of ischemia (e.g., arrested forearm circulation) or a sudden elevation in blood pressure (cold pressor test; see Chapters 49 and 58). Mounting evidence indicates that smoldering vascular inflammation contributes to the genesis and complications of high blood pressure. C-reactive protein (CRP) (see Chapters 10 and 42), an easily measured serum biomarker, “reports” on inflammation.40 Cross-sectional studies show strong correlations between elevated CRP and arterial stiffness and elevated pulse pressure. Longitudinal studies implicate elevated CRP levels as a risk marker for new onset of hypertension and accelerated progression of hypertensive target organ disease, possibly beyond that explained by blood pressure elevation alone. Oxidative stress also contributes to endothelial cell vasodilator dysfunction in hypertension. Superoxide anion and other reactive oxygen species quench nitric oxide, thereby reducing its bioavailability.41 Several pathways produce superoxide in arteries: nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, which are expressed in all vascular cell types and activated by circulating A II; nitric oxide synthase, which produces superoxide only when an important cofactor (tetrahydrobiopterin) is deficient, a process known as nitric oxide synthase uncoupling; xanthine oxidase, which produces uric acid; and mitochondria. Generation of reactive oxygen species by xanthine oxidase accounts for the association of hyperuricemia with endothelial dysfunction and hypertension. The xanthine oxidase inhibitor allopurinol can normalize blood pressure in two thirds of adolescents with hyperuricemia and recently diagnosed hypertension and can rectify prehypertension in obese adolescents42 but cannot be recommended as a routine antioxidant because of its serious side effect profile. The weak antioxidant vitamins C and E have little effect on blood pressure. Over time, endothelial cell dysfunction, neurohormonal activation, and elevated blood pressure cause remodeling of blood vessels, which further perpetuates hypertension43 (Fig. 43-9). An increase in the medial thickness relative to the lumen diameter (increased media-to-lumen ratio) is the hallmark of hypertensive remodeling in small and large arteries. Vasoconstriction initiates small-artery remodeling, which normalizes wall stress. Normal smooth muscle cells (SMCs) rearrange themselves around a smaller lumen diameter, a process termed inward eutrophic remodeling. The media-to-lumen ratio increases, but the medial cross-sectional area remains unchanged. By decreasing lumen diameter in the peripheral circulation, inward eutrophic remodeling increases systemic vascular resistance, the hemodynamic hallmark of diastolic hypertension. In contrast, large-artery remodeling is characterized by the expression of hypertrophic genes, triggering increases in medial thickness and in the media-to-lumen ratio. Such hypertrophic remodeling involves an increase in the size of vascular SMCs and an accumulation of extracellular matrix proteins, such as collagen, due to activation of transforming growth factor-beta (TGF-β). The resultant large-artery stiffness is the hemodynamic hallmark of ISH. Antihypertensive therapy may not provide optimal cardiovascular protection unless it prevents or reverses vascular remodeling by normalizing hemodynamic load, restoring normal endothelial cell function, and eliminating the underlying neurohormonal activation.43 Activation of the renin-angiotensin-aldosterone system (RAAS) (see Fig. 22-4) is one of the most important mechanisms contributing to endothelial cell dysfunction, vascular remodeling, and hypertension (Fig. 43-10). Renin, a protease produced solely by the renal juxtaglomerular cells, cleaves angiotensinogen (renin substrate produced by the liver) to angiotensin I (A I), which is converted to A II by angiotensin-converting enzyme (ACE) (see Chapter 88). ACE is most abundant in the lungs but also is present in the heart and systemic vasculature (tissue ACE). Chymase, a serine protease in the heart and systemic arteries, provides an alternative pathway for conversion of A I to A II. The interaction of A II with G protein–coupled AT1 receptors activates numerous cellular processes that contribute to hypertension and accelerate hypertensive end-organ damage (see Fig. 43-10), including vasoconstriction, generation of reactive oxygen species, vascular inflammation, vascular and cardiac remodeling, and production of aldosterone, the principal mineralocorticoid. Increasing evidence shows that that aldosterone, A II, and even renin and prorenin activate multiple signaling pathways that can damage vascular health and cause hypertension.
Systemic Hypertension
Mechanisms and Diagnosis
Definition, Prevalence, Variability, and Determinants of Hypertension
Definition
Prevalence
Blood Pressure Variability and Its Determinants
Behavioral Determinants
Genetic Determinants
Mechanisms of Primary (Essential) Hypertension
Hemodynamic Subtypes
Systolic Hypertension in Teenagers and Young Adults
Diastolic Hypertension in Middle Age
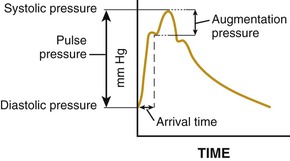
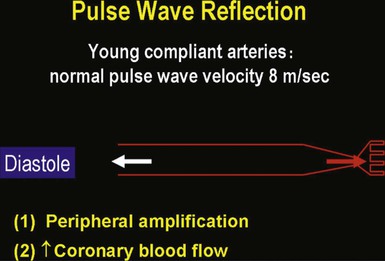
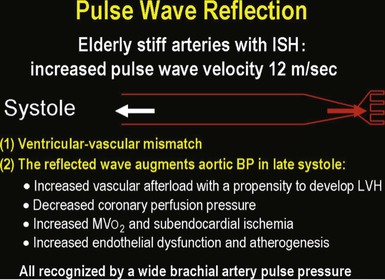
Isolated Systolic Hypertension in Older Adults


 ).17 Accumulation of collagen (which is poorly distensible) adversely affects its ratio to elastin in the aortic wall.
).17 Accumulation of collagen (which is poorly distensible) adversely affects its ratio to elastin in the aortic wall.
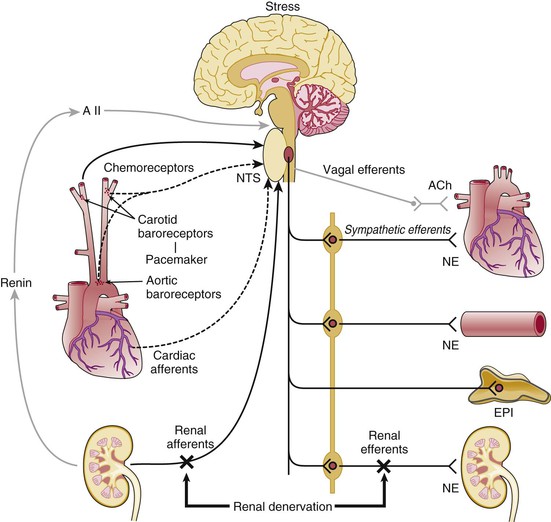
Neural Mechanisms
The Carotid Baroreceptor Pacemaker
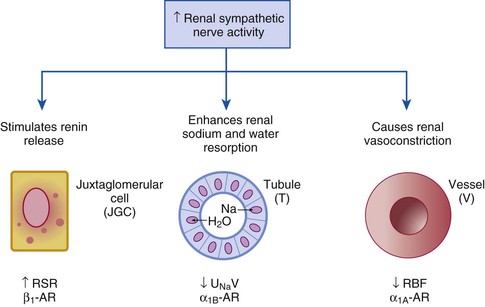
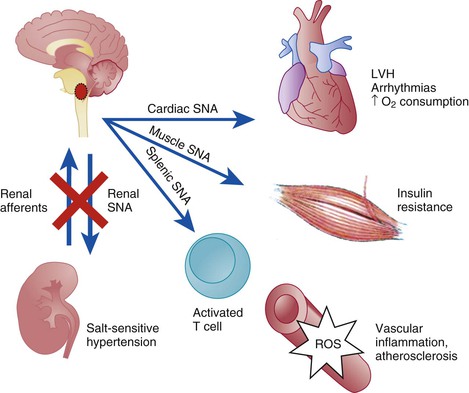
Catheter-Based Renal Denervation
Obesity-Related Hypertension
Obstructive Sleep Apnea as a Cause of Neurogenic Hypertension
Renal Mechanisms
Resetting of Pressure-Natriuresis
Low Birth Weight
Genetic Contributions
Vascular Mechanisms
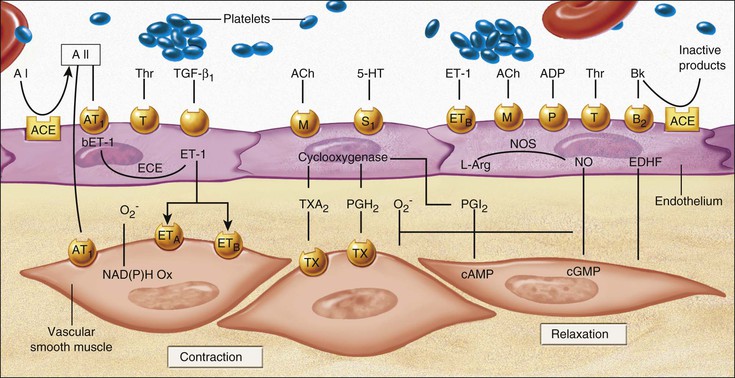
Endothelial Cell Dysfunction
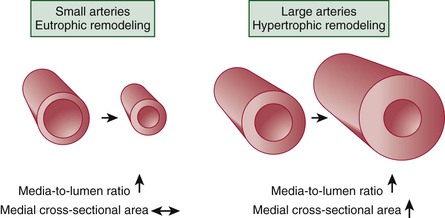
Vascular Remodeling
Hormonal Mechanisms
Renin-Angiotensin-Aldosterone System