William J. Groh, Douglas P. Zipes
Neurologic Disorders and Cardiovascular Disease
Cardiologists are increasingly being asked to evaluate and treat patients with a primary neurologic disorder because of the potential for associated cardiac involvement. In several neurologic disorders, the cardiovascular manifestations are responsible for a greater proportion of morbidity and mortality risk than that attributable to the neurologic manifestations. This chapter reviews those neurologic disorders associated with important cardiovascular manifestations or sequelae.
The Muscular Dystrophies
The muscular dystrophies are a group of inherited skeletal muscle diseases. Most also have direct effects on cardiac muscle, with manifestations including heart failure, conduction disease and heart block, atrial and ventricular arrhythmias, and sudden death. With improved multidisciplinary care, patients are living longer, with increased numbers exhibiting associated cardiac-related issues. As discussed next individually, the following muscular dystrophies can manifest with cardiovascular involvement:
• Duchenne and Becker muscular dystrophies
• Emery-Dreifuss muscular dystrophies and associated disorders
• Limb-girdle muscular dystrophies
Duchenne and Becker Muscular Dystrophies
Clinical Presentation
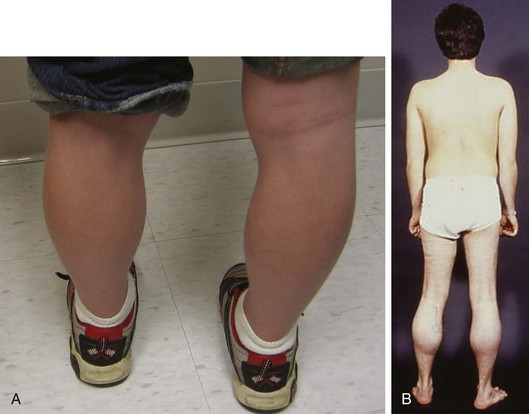
Duchenne muscular dystrophy is the most common inherited neuromuscular disease with an incidence of 1 case in 3600 to 6000 live male births.3 Patients typically present with skeletal muscle weakness before the age of 5 years, which progresses if untreated such that boys become wheelchair-bound by their early teens (Fig. 87-1). Historically, death occurs by age 25 years, primarily from respiratory dysfunction and less often from heart failure. A multidisciplinary treatment approach including steroids, scoliosis surgery, ventilatory support, and cardiac therapy has improved survival. Becker muscular dystrophy is less common than Duchenne muscular dystrophy, is associated with a more variable presentation of skeletal muscle weakness (see Fig. 87-1), and carries a better prognosis, with most patients surviving to the age of 40 to 50 years.
In both Duchenne and Becker muscular dystrophy, elevated serum creatine kinase activity is observed, at levels more than 10 and 5 times normal values, respectively.
Cardiovascular Manifestations
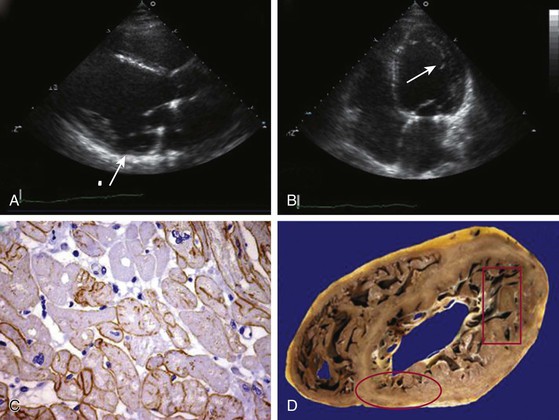
Most patients with Duchenne muscular dystrophy develop a cardiomyopathy, but symptoms can be masked by severe skeletal muscle weakness. Preclinical cardiac involvement is present in one fourth by age 6, with the onset of clinically apparent cardiomyopathy after age 10 common. Up to 90% of patients with Duchenne muscular dystrophy 18 years of age or older develop a dilated cardiomyopathy. Predilection for involvement in the inferobasal and lateral left ventricle has been observed (Fig. 87-2). As with the skeletal muscle weakness, cardiac involvement in Becker muscular dystrophy is more variable than in Duchenne muscular dystrophy, ranging from none or subclinical to severe cardiomyopathy requiring transplant. Cardiac involvement in Becker muscular dystrophy is independent of the severity of skeletal muscle involvement, with some but not all investigators observing increased likelihood of cardiovascular disease in older patients. More than one half of patients with subclinical or benign skeletal muscle disease were noted to have cardiac involvement if carefully evaluated. Progression in the severity of cardiac involvement is common. Cardiomyopathy can initially solely involve the right ventricle.
Thoracic deformities and a high diaphragm can alter the cardiovascular examination in patients with Duchenne muscular dystrophy. A reduction in the anterior-posterior chest dimension is commonly responsible for a systolic impulse displaced to the left sternal border, a grade 1 to 3/6 short midsystolic murmur in the second left interspace and a loud pulmonary component of the second heart sound. In both Duchenne and Becker types of muscular dystrophy, mitral regurgitation is observed. The presence of mitral regurgitation is related to posterior papillary muscle dysfunction in Duchenne muscular dystrophy and to mitral annular dilation in Becker muscular dystrophy.
Female carriers of Duchenne and Becker muscular dystrophy are at increased risk for dilated cardiomyopathy.
Electrocardiography
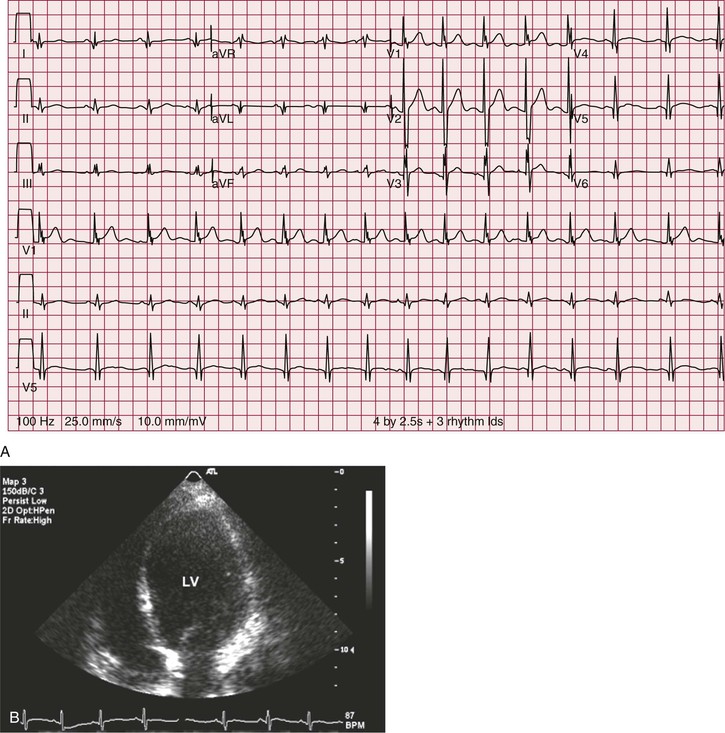
In a majority of patients with Duchenne muscular dystrophy, the electrocardiographic tracing is abnormal (see Chapter 12). The classically described electrocardiographic pattern shows distinctive tall R waves and increased R/S amplitude in V1 and deep narrow Q waves in the left precordial leads possibly related to the posterolateral left ventricular involvement (Fig. 87-3). Other common findings include short PR interval and right ventricular hypertrophy. No association between the presence of a dilated cardiomyopathy and electrocardiographic abnormalities has been established.4 In Becker muscular dystrophy, electrocardiographic abnormalities are present in up to 75% of the patients. The electrocardiographic abnormalities observed include tall R waves and increased R/S amplitude in V1, akin to that seen in Duchenne muscular dystrophy. Incomplete right bundle branch block also is a frequent finding that may be related to early involvement of the right ventricle. In patients with dilated cardiomyopathy, a left bundle branch block is common.
Echocardiography and Other Imaging Modalities
Clinical care guidelines recommend screening echocardiography at diagnosis or by the age 6 of years and subsequently every 2 years until the age of 10 and annually thereafter in boys with Duchenne muscular dystrophy3 (this and other cardiac imaging modalities are described more fully in Chapters 14 to 18). Abnormalities in strain imaging, diastolic dysfunction, and regional wall motion abnormalities can precede global systolic dysfunction in both Duchenne and Becker muscular dystrophy.5 Regional abnormalities in the posterobasal and lateral wall typically occur earlier than in other areas (see Fig. 87-2). A process akin to left ventricular noncompaction can be observed, possibly resulting from compensatory mechanisms in response to the failing dystrophic myocardium. Mitral regurgitation can result from dystrophic changes in the posterior leaflet papillary muscles. Cardiac magnetic resonance imaging is more sensitive in detecting subclinical ventricular involvement and fibrosis.
Arrhythmias
In Duchenne muscular dystrophy, persistent or labile sinus tachycardia is the most common arrhythmia recognized (see Chapter 37). Atrial arrhythmias including atrial fibrillation and atrial flutter occur in the setting of respiratory dysfunction and cor pulmonale or are associated with a dilated cardiomyopathy. Abnormalities in atrioventricular conduction have been observed, with both short and prolonged PR intervals recognized. Ventricular arrhythmias occur on monitoring in 30%, primarily ventricular premature beats. Complex ventricular arrhythmias have been reported, more commonly in patients with severe skeletal muscle disease. Sudden death occurs in Duchenne muscular dystrophy, typically in patients with end-stage muscular disease. Whether the sudden death is caused by arrhythmias is unclear. Several follow-up studies have shown a correlation between sudden death and the presence of complex ventricular arrhythmias. The presence of ventricular arrhythmias was not a predictor for all-cause mortality.
Arrhythmia manifestations in Becker muscular dystrophy typically relate to the severity of the associated structural cardiomyopathy. Distal conduction system disease with complete heart block and bundle branch reentry ventricular tachycardia has been observed.
Treatment and Prognosis
Duchenne muscular dystrophy is a progressive skeletal and cardiac muscle disorder. Steroids and steroid derivatives are effective in delaying skeletal muscle disease progression and also appear to decrease the progression to a dilated cardiomyopathy. Drisapersen is an antisense oligonucleotide that facilitates “exon skipping” of a nonsense mutation in the dystrophin gene, which creates a premature stop signal in the translation of the dystrophin mRNA, thereby leading to a prematurely truncated, dysfunctional dystrophin protein. Nonsense mutations of dystrophin genes are responsible for dystrophinopathy in approximately 10% to 15% of boys with the disease. Oligonucleotide-mediated exon skipping restores the open reading frame of the dystrophin gene, resulting in an internally deleted (truncated) but semi-functional dystrophin. Early phase clinical trials have demonstrated clinical benefit with an acceptable safety profile, and the FDA has given drisapersen favorable guidance for a regulatory path forward, under an accelerated approval pathway. Gene replacement therapy also holds future promise. A primary cardiac cause of death is recognized to play an increasingly significant role because of delayed mortality with improved respiratory support. There is an equal distribution of cardiac death from heart failure and sudden death. Angiotensin-converting enzyme inhibitors and beta blockers can improve left ventricular function in patients treated early.1,2 Spironolactone may provide additional benefit (see Chapters 24 and 25).6 Other advanced therapy such as implantable cardioverter-defibrillators are of uncertain role but should be considered individually based on patient status and wishes (see Chapter 36). Whether heart failure therapies improve long-term outcomes is unclear. Age at death has increased so that the majority of patients survive into their 30s.
In patients with Becker muscular dystrophy, an improvement in left ventricular function also is observed after treatment with angiotensin-converting enzyme (ACE) inhibitors and beta blockers.1 Screening left ventricular imaging is recommended as in Duchenne muscular dystrophy. Advanced heart failure therapy, including primary prevention implantable cardioverter-defibrillators, is appropriate in patients with cardiomyopathy. Patients with Becker muscular dystrophy with advanced heart failure can undergo cardiac transplantation, with expected outcomes similar to those for non–muscular dystrophy cohorts of age-matched patients with dilated cardiomyopathy.7 Female carriers of Duchenne and Becker muscular dystrophies do not develop a cardiomyopathy during childhood, and screening can be delayed until later in adolescence. Whether carriers benefit from afterload reduction therapy is unknown, but such treatment would seem reasonable based on shared mechanisms. Once heart failure is established, conventional therapy is indicated. Cardiac transplantation also has been reported in carriers.
Myotonic Dystrophies
Genetics.
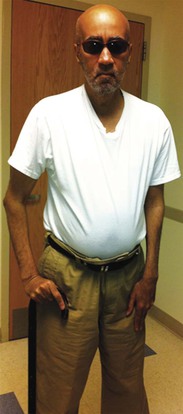
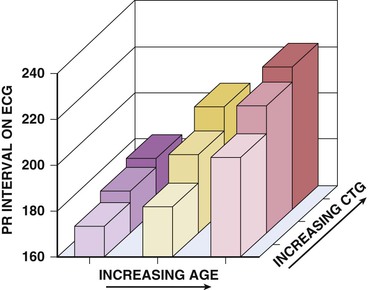
The myotonic dystrophies are autosomal dominant disorders characterized by myotonia, which is a delayed muscle relaxation after contraction, weakness and atrophy of skeletal muscles, and systemic manifestations including endocrine abnormalities, cataracts, cognitive impairment, and cardiac involvement (Fig. 87-4). Two distinct mutations are responsible for the myotonic dystrophies. In myotonic dystrophy type 1, the mutation is an amplified trinucleotide cytosine-thymine-guanine (CTG) repeat found on chromosome 19. Whereas unaffected patients have 5 to 37 copies of the repeat, patients with myotonic dystrophy have 50 to several thousand repeats. A direct correlation exists between an increasing number of CTG repeats and earlier age at onset and increasing severity of neuromuscular involvement. Cardiac involvement including conduction disease and arrhythmias also correlate with the length of repeat expansion (Fig. 87-5). It is typical for the CTG repeat to expand as it is passed from parents to offspring, resulting in the characteristic worsening clinical manifestations in subsequent generations, termed anticipation.
Myotonic dystrophy type 2, also called proximal myotonic myopathy, has generally less severe skeletal muscle and cardiac involvement than in type 1. Both congenital presentation and cognitive impairment are lacking in myotonic dystrophy type 2—typically the most severely involved subsets of the type 1 patients. The genetic mutation responsible for myotonic dystrophy type 2 is a tetranucleotide repeat expansion, cytosine-cytosine-thymine-guanine (CCTG), found on chromosome 3. Intergenerational contraction of the repeat expansion has been reported, and there is no apparent relationship between the degree of expansion and clinical severity.
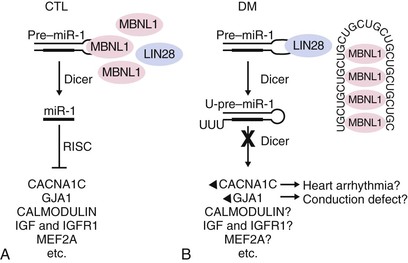
The molecular mechanism by which both myotonic dystrophies exert their similar phenotypic presentations is by the toxic effects of the large mutant RNA expansion on nuclear RNA binding proteins.8 A recent study suggests that cardiac pathology in both myotonic dystrophy type 1 and 2 is related to gap junction (GJA1) and calcium channel (CACNA1C) protein overexpression9 (Fig. 87-6).
Clinical Presentation
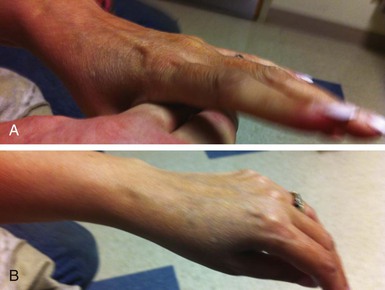
The myotonic dystrophies are the most common inherited neuromuscular disorders in patients presenting as adults. Until recently, studies have not genetically differentiated myotonic dystrophy types 1 and 2, so the clinical characteristics described probably are for a mixed group of such disorders. Type 1 is significantly more common than type 2, except possibly in certain areas of northern Europe. The global incidence of myotonic dystrophy type 1 has been estimated to be 1 in 8000, although it is higher in certain populations, such as French Canadians, and lower to nonexistent in other populations, such as African blacks. The age at onset of symptoms and diagnosis averages 20 to 25 years. A congenital presentation is seen in severely affected patients with myotonic dystrophy type 1. Common early manifestations are related to weakness in the muscles of the face, neck, and distal extremities. On examination, myotonia can be demonstrated in the grip, thenar muscle group, and tongue (Fig. 87-7). Diagnosis when the patient is asymptomatic is possible using electromyography and genetic testing. Muscle weakness is progressive. Subcapsular cataracts are commonly observed. In general, cardiac symptoms appear after the onset of skeletal muscle weakness but can be the initial manifestation of the disease.
Myotonic dystrophy type 2 also manifests with myotonia, muscle weakness, cataracts, and endocrine abnormalities, as in type 1. Age at symptom onset typically is older in myotonic dystrophy type 2.
Cardiovascular Manifestations
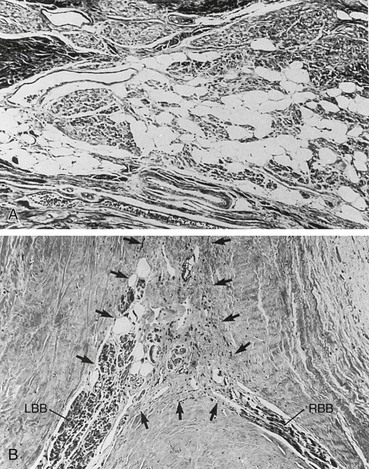
Cardiac pathology in the myotonic dystrophies involves degeneration, fibrosis, and fatty infiltration preferentially targeting the specialized conduction tissue including the sinus node, atrioventricular node, and His-Purkinje system (Fig. 87-8). Degenerative changes are observed in working atrial and ventricular tissue but only rarely progress to a symptomatic dilated cardiomyopathy. It is not clear if there are differences in the cardiac pathology observed between myotonic dystrophy type 1 and 2. Patients with type 2 typically demonstrate cardiac involvement later in life or not at all. The primary cardiac manifestations of the myotonic dystrophies are arrhythmias.
Electrocardiography
A majority of adult patients with myotonic dystrophy type 1 exhibit electrocardiographic abnormalities. In a large, unselected middle-aged U.S. myotonic population, abnormal electrocardiographic patterns were seen in 65% of the patients.10 Abnormalities included first-degree atrioventricular block in 42%, right bundle branch block in 3%, left bundle branch block in 4%, and nonspecific intraventricular conduction delay in 12%. Q waves not associated with a known myocardial infarction are common. Electrocardiographic abnormalities are less common in younger patients. Conduction disease worsens with advancing age (Fig. 87-9).
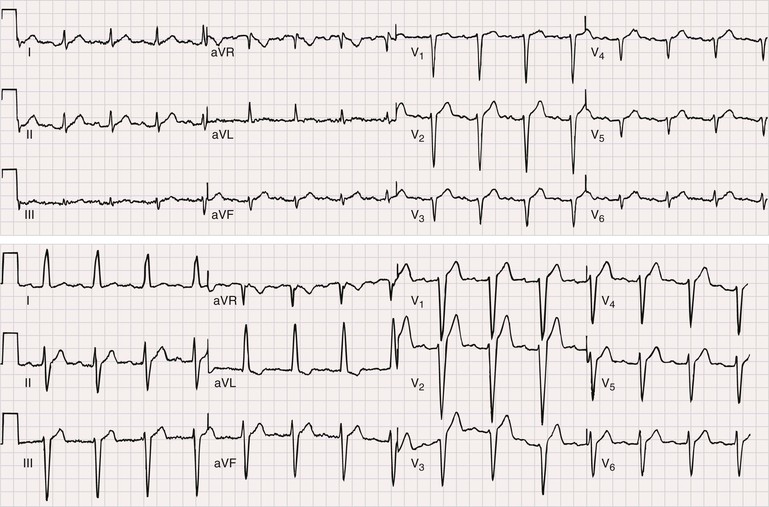
Electrocardiographic abnormalities are less common in myotonic dystrophy type 2, occurring in approximately 20% of middle-aged patients.
Echocardiography and Other Imaging Modalities
Left ventricular systolic and diastolic dysfunction, left ventricular hypertrophy, mitral valve prolapse, regional wall motion abnormalities, and left atrial dilatation have been reported in patients with myotonic dystrophy type 1 at moderate prevalence rates.11 Clinical heart failure is observed but is less common than are arrhythmias. Left ventricular hypertrophy and ventricular dilation have been reported in myotonic dystrophy type 2. Cardiac magnetic resonance imaging may be more sensitive than echocardiography for detection of early cardiac involvement.
Arrhythmias
Patients with myotonic dystrophy type 1 demonstrate a wide range of arrhythmias. At cardiac electrophysiologic study, the most common abnormality found is a prolonged His-ventricular (H-V) interval. Conduction system disease can progress to symptomatic atrioventricular block and necessitate pacemaker implantation. The prevalence of permanent cardiac pacing in patients with myotonic dystrophy type 1 varies widely between studies based on referral patterns and the indications used for implant. Updated practice guidelines have recognized that asymptomatic conduction abnormalities in neuromuscular diseases such as myotonic dystrophy may warrant special consideration for pacing.12
Atrial arrhythmias, primarily atrial fibrillation and atrial flutter (see Chapter 38), are the most common arrhythmias observed.10 Ventricular tachycardia can occur. Patients with myotonic dystrophy type 1 are at risk for ventricular tachycardia occurring as a consequence of reentry in the diseased distal conduction system, as characterized by bundle branch reentry and interfascicular reentry tachycardia (Fig. 87-10). Therapy with right bundle branch or fascicular radiofrequency ablation can be curative.
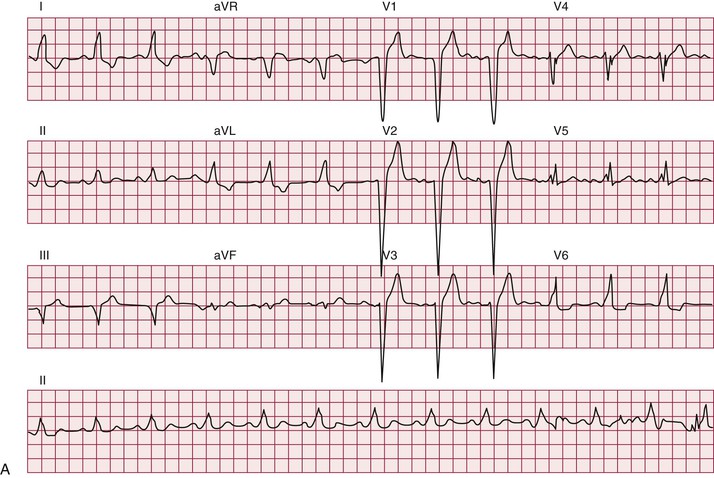
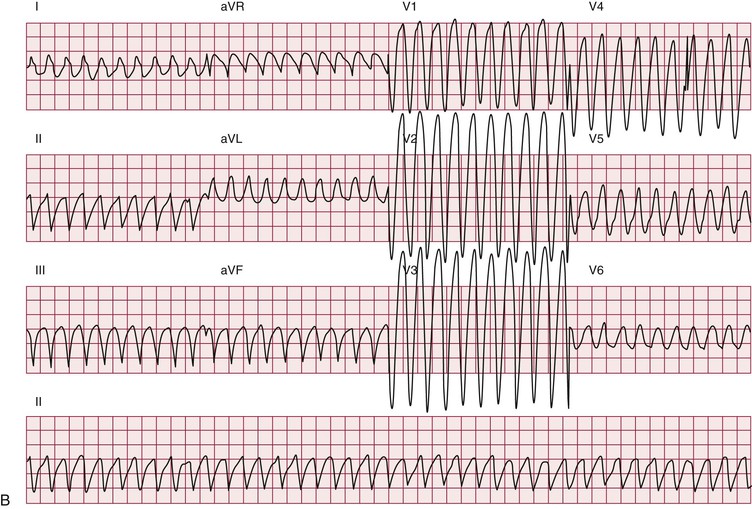
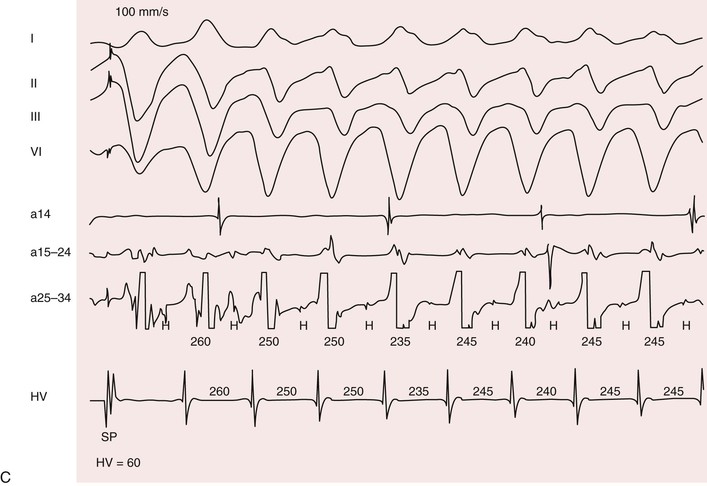
Up to one third of deaths in myotonic dystrophy type 1 are sudden; presumably, most are due to arrhythmias.10 The entity of sudden death is second only to respiratory failure as a cause of death. The mechanisms leading to sudden death are not clear. Distal conduction disease producing atrioventricular block can result in the lack of an appropriate escape rhythm and asystole or bradycardia-mediated ventricular fibrillation. Sudden death can occur in myotonic dystrophy type 1 despite pacing, implicating ventricular arrhythmias. Nonarrhythmia causes of sudden death, probably acute respiratory issues, play some role.
Arrhythmias and sudden death have been reported in myotonic dystrophy type 2 but seem to be rarer than in type 1.
Treatment and Prognosis
Neurologists recognize the risk for cardiac issues in the myotonic dystrophies and will refer to cardiology. Cardiac manifestations occur in both myotonic dystrophy types 1 and 2 and therefore diagnostic evaluation and therapy should be done in both. Cardiac disease is observed at a younger age in myotonic dystrophy type 1 compared with type 2. Echocardiography or other imaging modalities can determine if structural abnormalities are present. Cardiac imaging in adults should be done at diagnosis or with new symptoms. If no significant abnormalities are observed repeat evaluation every 3 to 5 years is appropriate. In the patient with a dilated cardiomyopathy, standard therapy including ACE inhibitors and beta blockers has improved symptoms. There are no data on the role of ACE inhibitors or beta blockers to prevent development of a cardiomyopathy in myotonic dystrophy. Patients presenting with symptoms indicative of arrhythmias such as syncope and palpitations should undergo an evaluation, often including a cardiac electrophysiologic study, to determine an underlying causative disorder. Annual electrocardiograms (ECGs) are recommended in asymptomatic patients. The role and interval for ambulatory ECG (Holter) monitoring are not clear (see Chapter 34). Significant or progressive electrocardiographic abnormalities despite a lack of symptoms is an indication for consideration for prophylactic pacing.12 The presence of severe electrocardiographic conduction abnormalities and atrial arrhythmias were independent risk factors for sudden death.10
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


