CHAPTER 67 Surgery of the Aortic Root and Ascending Aorta
FUNCTIONAL ANATOMY OF THE AORTIC ROOT
The subcommissural triangles are part of the left ventricular outflow tract, but they play an important role in the function of the aortic valve. The subcommissural triangles of the noncoronary aortic cusp are fibrous extension of the intervalvular fibrous body and membranous septum, whereas the subcommissural triangle beneath the left and the right aortic cusps is an extension of the muscular interventricular septum (Fig. 67-1). The aortic anulus, a fibrous structure with a scalloped shape, attaches the aortic valve to the left ventricle. It is attached directly to the myocardium in approximately 45% of its circumference, and to fibrous structures in the remaining 55%. Histologic examination of the aortic anulus reveals that the aortic root has a fibrous continuity with the anterior leaflet of the mitral valve and membranous septum, and it is attached to the muscular interventricular septum by fibrous strands (Fig. 67-2). An important structure immediately below the membranous septum is the bundle of His. The atrioventricular node lies in the floor of the right atrium between the tricuspid anulus and the coronary sinus orifice. This node gives origin to the bundle of His, which travels through the right fibrous trigone along the posterior edge of the membranous septum to the muscular interventricular septum. At this point, the bundle of His divides into left and right bundle branches, which run subendocardially along both sides of the interventricular septum.
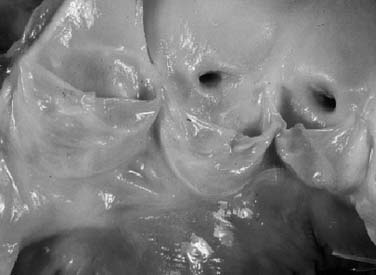
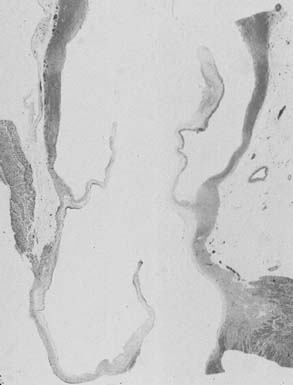
The normal aortic root has a fairly consistent shape, and the sizes of the cusps, the aortic anulus, the aortic sinuses, and the sinotubular junction are somewhat interdependent.1–4 Thus, large cusps have a proportionally large anulus, sinus, and sinotubular junction. The three aortic cusps often have different sizes in a person, and the right and noncoronary cusps are usually larger than the left cusp.3 The same cusp may have different sizes in individuals with the same body surface area.3,4 There are, however, certain geometric parameters that are fairly constant among the various components of the aortic root, and this knowledge is indispensable to understanding the principles of aortic valve repair or replacement with stentless biological valves.
The free margin of an aortic cusp extends from one of its commissures to the other. The length of the free margin of an aortic cusp is approximately 1.5 times the length of its base (Fig. 67-3). During diastole, the free margins and part of the body of the three cusps touch each other approximately in the center of the aortic root to seal the aortic orifice. Thus, the average length of the free margins of three aortic cusps must exceed the diameter of the sinotubular junction to allow the cusps to coapt centrally and render the aortic valve competent. If a pathologic process causes shortening of the length of the free margin of a cusp, or if the sinotubular junction dilates, the cusps cannot coapt centrally, resulting in aortic insufficiency (Fig. 67-4). If the length of a free margin is elongated, the cusp prolapses, and depending on the degree of prolapse, aortic insufficiency ensues (Fig. 67-5).
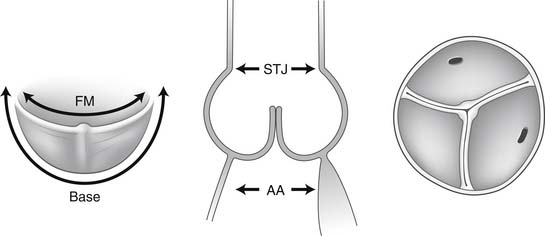
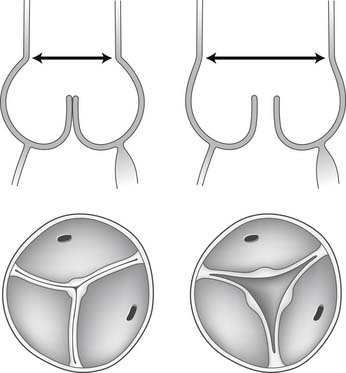
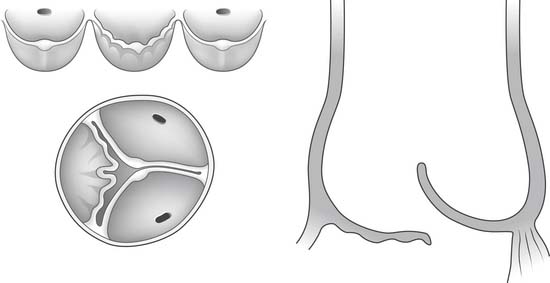
Figure 67–5 Elongation of the free margin of an aortic cusp causes prolapse with resulting aortic insufficiency.
The diameter of the aortic anulus is 10% to 20% larger than the diameter of the sinotubular junction of the aortic root in young patients (see Fig. 67-3). As the number of elastic fibers in the arterial wall decreases with age, the sinotubular junction dilates, and its diameter tends to become equal to that of the aortic anulus in older patients.
Dilation of the aortic anulus pulls the belly of the aortic cusps apart, decreasing the coaptation area, and it eventually causes aortic insufficiency (Fig. 67-6). With dilation of the aortic anulus, the subcommissural triangles of the noncoronary cusp tend to become more obtuse as the crescent shape of the aortic anulus along its fibrous insertion flattens (see Fig. 67-6). The subcommissural triangle beneath the right and left cusps does not change much in patients with annuloaortic ectasia because it is part of the muscular interventricular septum and is not affected by the connective tissue disorder that causes dilation of the fibrous skeleton of the heart.
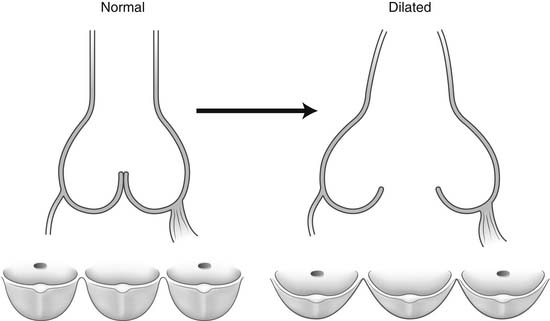
Figure 67–6 Dilation of the aortic anulus. The subcommissural triangles of the noncoronary cusp become more obtuse.
The aortic sinuses facilitate closure of the aortic valve by creating eddies and currents between the cusps and arterial wall (Fig. 67-7). They also prevent the cusps from occluding the coronary artery orifices during systole, thus guaranteeing myocardial perfusion during the entire cardiac cycle. Isolated dilation of the aortic sinuses does not cause aortic insufficiency.5
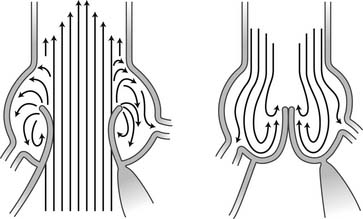
The aortic root of young individuals is elastic and very compliant. It expands and contracts during the cardiac cycle. Expansion and contraction of the aortic anulus are heterogeneous, probably because of its attachments to contractile myocardium and to fibrous structures such as the membranous septum and intervalvular fibrous body. On the other hand, the expansion and contraction of the sinotubular junction are more uniform. The aortic root also displays some degree of torsion during isovolumic contraction and ejection of the left ventricle.6 Compliance decreases with aging because of loss of elastic fibers, and the movements of the aortic anulus, cusps, sinuses, and sinotubular junction also change.
PATHOLOGY OF THE AORTIC ROOT AND ASCENDING AORTA
The wall of the aorta is composed of three layers: intima, media, and adventitia. The intima is a thin layer of ground substance lined by endothelium, and it is easily traumatized. The media is the thickest of the three layers, and it is made of elastic fibers arranged in spiral fashion to increase the tensile strength. The adventitia is a thin fibrous layer and contains the vasa vasorum, which carry the nutrients to the media. The aorta is very compliant and expands and contracts during the cardiac cycle because of the elastic fibers in the media. Compliance decreases with aging because of fragmentation of the elastic fibers and an increase in the amount of fibrous tissue in the media. Hypertension, hypercholesterolemia, and coronary artery disease cause premature aging of the aorta.7–9 Exercise seems to protect the elasticity of the aorta.8
Degenerative Aneurysm of the Ascending Aorta
Ascending aortic aneurysms tend to increase in size and eventually rupture or cause aortic dissection. The transverse diameter of the aneurysm is the most important predictor of rupture or dissection. In a study by Coady and associates10 of 370 patients with thoracic aneurysms (201 ascending aortic aneurysms), during a mean follow-up of 29.4 months, the incidence of acute dissection or rupture was 8.8% for aneurysms less than 4 cm, 9.5% for aneurysms of 4 to 4.9 cm, 17.8% for 5 to 5.9 cm, and 27.9% for those greater than 6 cm. The median size of the ascending aortic aneurysm at the time of rupture or dissection was 5.9 cm.
The growth rates of thoracic aneurysms are exponential.10 In the Coady study, the growth rate ranged from 0.08 cm/yr for small (<4-cm) aneurysms to 0.16 cm/yr for large (8-cm) aneurysms.10 The growth rates for chronic dissecting aneurysms were much higher than for chronic nondissecting aneurysms. Other studies found greater annual growth rates than the Coady group.11,12 In addition, the growth rates for aortic root aneurysms may be different from those of ascending aortic aneurysms.
Most patients with aortic root or ascending aortic aneurysms are asymptomatic, and the aneurysm is usually found during routine chest radiographs, which show a widened mediastinum.13 Tracheal and esophageal displacement may be observed in the posterolateral view of the chest radiographs. Approximately one third of the patients complain of vague chest pain.13 In patients with a massive ascending aortic aneurysm, signs of superior vena cava obstruction may be present. If aortic insufficiency is present, there may be cardiac enlargement and the physical findings associated with it. The diagnoses of aortic root and ascending aortic aneurysm can be confirmed by echocardiography.
Magnetic resonance imaging (MRI) provides even more information than CT scanning because it visualizes the arterial wall and surrounding structures with greater contrast. In addition, it has been increasingly used in the diagnosis and management of patients with heart diseases.14 Magnetic resonance angiography (MRA) is replacing contrast angiography.15
Marfan Syndrome
The clinical features of Marfan syndrome were thought to be caused by weaker connective tissues resulting from defects in fibrillin-1, a glycoprotein and principal component of the extracellular matrix microfibril. This concept was inadequate to explain the overgrowth of long bones, osteopenia, reduced muscular mass and adiposity and craniofacial abnormalities often seen in Marfan syndrome.16 Dietz and colleagues16,17 showed in experimental mice with Marfan syndrome that many findings are the result of abnormal levels of activation of transforming growth factor β (TGF-β), a potent stimulator of inflammation, fibrosis, and activation of certain matrix metalloproteinases, especially matrix metalloproteinases 2 and 9. Excess TGF-β activation in tissues correlates with failure of lung septation, development of a myxomatous mitral valve, and aortic root dilation in mice.18 This combination of structural microfibril matrix abnormality, dysregulation of matrix homeostasis mediated by excess TGF-β, and abnormal cell–matrix interactions is responsible for the phenotypic features of Marfan syndrome.16–18 Ongoing destruction of the elastic and collagen lamellae and medial degeneration result in progressive dilation of the aortic root, as well as in a predisposition to aortic dissection from the loss of appropriate medial layer support. Loss of elasticity in the media causes increased aortic stiffness and decreased distensibility.19
The diagnosis of Marfan syndrome is made on clinical grounds, and it is not always simple because of the variability in clinical expression (Table 67-1). A multidisciplinary approach is needed to diagnose and manage patients afflicted with this syndrome. The presence of major criteria in two separate systems and involvement of a third (minor or major) are needed to establish the diagnosis.20
Table 67–1 Diagnostic Criteria∗ for Marfan Syndrome (See text)
| System | Major Criteria | Minor Criteria |
|---|---|---|
| Family history | Independent diagnosis in parent, child, sibling | None |
| Genetics | Mutation in FBN1 | None |
| Cardiovascular | ||
| Ocular | Ectopia lentis | Two needed: |
| Skeletal | Two major or one major and two minor signs: | |
| Pulmonary | — | |
| Skin | — | |
| Central nervous | Lumbosacral dural ectasia (CT or MRI) | — |
∗ The presence of major criteria in two separate systems and involvement of a third (minor or major) are needed to establish the diagnosis of Marfan syndrome.
Mitral valve prolapse is age dependent and more common in women. It is caused by myxomatous degeneration of the mitral valve apparatus, which is present in up to 80% of patients with Marfan syndrome, but only 25% of them develop mitral insufficiency. The posterior mitral anulus is grossly dilated in patients with mitral insufficiency, and it is often displaced posteriorly.21 The mitral anulus may also become heavily calcified and display a horseshoe appearance on radiographs.
The dilation of the aortic root is often progressive, and the rate of expansion, which varies somewhat, is usually less than 1 or 2 mm/yr. Shores and colleagues12 randomized 70 patients with Marfan syndrome into propranolol-treated and placebo groups. The growth rates of the aortic root aneurysms in untreated patients were slightly more than three times those of patients who received β-adrenergic blockage. This study has been the scientific basis to treat these patients with a beta-blocker agent. Calcium antagonists and angiotension-converting enzymes have also been reported as effective in delaying aortic dilation, but at present, beta-blocker remains the drug of choice.22
Aortic dissection is rare in patients with aortic root aneurysm of less than 50 mm, unless they have a family history of aortic dissection. The dissection in most patients starts at the level of the sinotubular junction (Stanford type A aortic dissection). In approximately 10% of patients, it starts just beyond the left subclavian artery (Stanford type B aortic dissection). Without surgery, most patients with Marfan syndrome die in their third decade, from complications of aortic root aneurysm such as rupture, aortic dissection, or aortic insufficiency.23,24
Pregnancy in women with Marfan syndrome has two potential problems: the risk of having a child who will inherit the disorder and the risk of acute aortic dissection during the third trimester, parturition, or the first postpartum month. Offspring have a 50% risk of inheriting the syndrome. The risk of aortic dissection is less known, but it appears to be low in patients with normal aortic root and cardiac function.25
Loeys-Dietz syndrome
Mutations in the genes encoding TGF-β receptors 1 and 2 have been found in association with a continuum of clinical features. On the mild end is a presentation similar to that of the Marfan syndrome, or familial thoracic aneurysm and dissection,26,27 and on the severe end, there is a complex phenotype in which aortic dissection or rupture commonly occurs in childhood.28 This complex phenotype is characterized by the triad of hypertelorism, bifid uvula (or cleft palate), and generalized arterial tortuosity with widespread vascular aneurysm and dissection. This phenotype has been classified as Loeys-Dietz syndrome.29 Affected patients have a high risk of aortic dissection or rupture at an early age and at relatively small aortic diameters. CT angiograms should be obtained from the head to the pelvis. Surgery is recommended for adults when the aortic root exceeds 4 cm or the descending thoracic aorta exceeds 5 cm. If the craniofacial features are severe in children, surgery is recommended when the aortic root Z-score is greater than 3, or when the expansion is greater than 0.5 cm in 1 year.30
Ehlers-Danlos Syndrome
The Ehlers-Danlos syndrome encompasses a group of heterogeneous connective tissue disorders that involve the skin and joints and cause hyperelasticity and fragility of the former and hypermobility of the latter. It may also involve the cardiovascular system. Vascular Ehlers-Danlos syndrome is a rare autosomal dominant inherited disorder of the connective tissue resulting from mutation of the COL3A1 gene encoding type III collagen.31 Affected individuals are prone to serious vascular, intestinal, and obstetric complications. These problems are rare during infancy but occur in up to 25% of affected persons before the age of 20 years and in 80% before the age of 40. Median survival is 48 years. Spontaneous rupture without dissection of large- and medium-caliber arteries such as the abdominal aorta and its branches, the branches of the aortic arch, and the large arteries of the limbs accounts for most deaths. Intestinal perforation, usually involving the colon, is less often fatal. Pregnancy is a high risk for women with this syndrome. Aortic root dilation was present in 28% in a series of 71 patients with Ehlers-Danlos syndrome.32 Aortic dissection is uncommon.
Bicuspid Aortic Valve Disease
Congenital aortic valve malformations include a phenotypic continuum of unicuspid valves (severe form), the various types of bicuspid aortic valves (moderate form), tricuspid valves (normal), and the rare quadricuspid valves.33
Bicuspid aortic valve, the most common of these malformations, occurs in 1% to 2% of the population. Movahed and colleagues34 recently reviewed 24,265 patients who had echocardiograms performed for various clinical reasons, and 1742 echocardiograms obtained by screening teenage athletes in Southern California, and found a prevalence of bicuspid aortic valves of 0.6% in the large cohort and 0.5% in the smaller one. Males are affected more than females at a ratio of 4:1. There is a relatively high incidence of familial clustering, which suggests an autosomal dominant inheritance with reduced penetrance.35,36 However, it remains unproved that bicuspid aortic valve is an inherited disorder. Patients with bicuspid aortic valve usually have three aortic sinuses and two cusps of different sizes. The larger cusp, usually the one attached to the interventricular septum, contains a raphe, which probably represents an incomplete commissure. Bicuspid aortic valves with two cusps and two sinuses are far less common than bicuspid aortic valves with two cusps and three sinuses. Most patients with bicuspid aortic valves have a dominant circumflex artery and a small right coronary artery. Normally functioning bicuspid aortic valves may last the patient’s lifetime. Others become stenotic by the fourth or fifth decade of life. Aortic insufficiency may also occur, and it is often associated with dilated aortic anulus.37 It is more common in younger patients, and it results from the prolapse of one cusp, usually the one that contains the raphe.
Both bicuspid and unicuspid aortic valves are often associated with premature degenerative changes in the media of the wall of the aortic root and ascending aorta.38,39 These patients are at risk of developing chronic degenerative aneurysms of the ascending aorta and type A aortic dissection.40
Atherosclerosis
Atherosclerosis of the ascending aorta and transverse arch is a common cause of stroke.41,42 Sometimes, atherosclerosis can cause extensive calcification of the aortic root, ascending aorta, and transverse arch, which is often associated with coronary artery disease, stenosis of one or both coronary arteries orifices, and aortic valve stenosis. Extensive calcification of the ascending aorta is clinically described as “porcelain” aorta.43,44
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


