CHAPTER 126 Surgery for Congenitally Corrected Transposition of the Great Arteries
Congenitally corrected transposition of the great arteries is a rare defect, seen in approximately 0.5% of patients with congenital heart defects.1,2 It entails discordance of the atrioventricular (AV) connections and discordance of the ventricular arterial connections, so there is double discordance. The morphology of the heart is distinctly abnormal, but the circulatory system is physiologically normal. Commonly, congenitally corrected transposition is associated with ventricular septal defects (VSDs), pulmonary stenosis, dysplasia of the tricuspid valve, and conduction abnormalities (e.g., heart block usually develops).3–5 Although this condition can be compatible with a normal life span,6,7 the majority of patients require surgery to repair the associated cardiac anomalies, and many develop heart failure because of systemic morphologic right ventricular (RV) dysfunction, usually with tricuspid valve regurgitation.8–11 Historically, surgery has been directed toward managing the associated cardiac anomalies, closing the VSD, relieving pulmonary stenosis, repairing or replacing the tricuspid valve, and placing pacemakers to manage the conduction abnormalities. This so-called classical or conventional (physiologic) repair maintains the morphologic right ventricle as the systemic ventricle. More recently, the morphologic left ventricle has been restored to the systemic circulation by an atrial switch (Senning or Mustard procedure) and an arterial switch or Rastelli procedure to connect the aorta to the morphologic left ventricle.12–15
GENERAL DESCRIPTION AND MORPHOLOGY
In the usual situs solitus arrangement with normal systemic and pulmonary venous drainage, the ventricular apex of the heart usually points to the left side of the patient, but mesocardia and dextrocardia are common, occurring in up to 20% of cases.4,16 Malposition of the ventricular mass so that it comes to lie in front of the atria and venous drainage to the heart can make surgical access to these structures difficult. In situs inversus, which occurs in about 5% of cases, mesocardia and levocardia can occur. It seems that extreme malposition of the ventricular mass is commonly associated with severe pulmonary stenosis or pulmonary atresia in association with a large VSD.16 Characteristically, the aorta is anterior and to the left side of the more deeply placed pulmonary artery—hence the older term for this condition, L-transposition. However, the aorta can be more anterior to the pulmonary artery and even on the right side. Likewise, the morphologic left ventricle is usually to the right and slightly inferior to the morphologic right ventricle; however, the position of the ventricles relative to each other can show great variability.3 The ascending aorta is often quite short and well over to the left side of the mediastinum in situs solitus, again making surgical access difficult. In the rarer form of situs inversus, the aorta lies anterior and to the right of the posterior pulmonary artery and is usually more accessible.
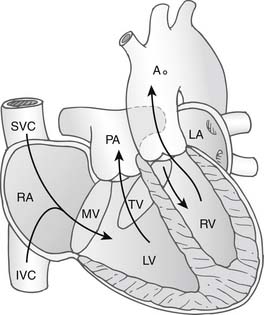
In congenitally corrected transposition of the great arteries without associated cardiac anomalies, the circulation is physiologically normal with systemic venous return passing through the right atrium, through the mitral valve, and then into the morphologic left ventricle. Here, the morphologic left ventricle pumps blood through the pulmonary arteries into the lungs, from whence it returns to the pulmonary veins and to the left atrium. The blood flow continues through the tricuspid valve and into the morphologic right ventricle, where it is pumped around the systemic circulation via the aorta (Fig. 126-1). Without other cardiac anomalies, patients with congenitally corrected transposition can survive well into adult life without symptoms. It is the associated anomalies—VSD, pulmonary stenosis, abnormalities of the tricuspid valve, and development of arrhythmias or complete heart block—that seem to predispose the patient to the development of cardiac failure early in life.
Ventricular Septal Defect
VSD occurs in about 70% of cases.4 It is usually isolated and in the perimembranous region. An isolated VSD can also occur in the infundibular region, and they may be multiple. When associated with severe pulmonary stenosis or pulmonary atresia, the VSD is usually subaortic and large, extending from the perimembranous region to beneath the aortic valve. This is important, because it allows the VSD to be tunneled to the aorta when the morphologic left ventricle is committed to the aorta as a systemic ventricle.
Pulmonary Outflow Tract Obstruction (Morphologic Left Ventricular Outflow Tract Obstruction)
Obstruction to the pulmonary artery from the left ventricle is common in congenitally corrected transposition, occurring in probably up to 50% of cases.4,17 This is partly because the pulmonary artery is between the mitral and tricuspid valves, deep in toward the crux of the heart. A potential exists for hypoplasia of the subpulmonary outflow tract. When this occurs in association with a VSD, there may be accessory tissue around the VSD, which can balloon into the pulmonary valve. Accessory tissue tags can prolapse into the outflow tract from either the mitral or the tricuspid valve, and over time fibrous tissue can be deposited in the subpulmonary area of the morphologic left ventricular (LV) outflow tract. Accessory attachments of the mitral or tricuspid mitral valve through the VSD can cause obstruction beneath the pulmonary valve. The pulmonary valve itself may be stenotic.
Mitral and Tricuspid Valves
The mitral valve is usually normal in congenitally corrected transposition. There are usually two well-formed papillary muscles on the lateral wall of the left ventricle. Occasionally, we have noted a cleft in the septal component of the mitral valve.18
The tricuspid valve, however, is a markedly different proposition. The valve itself may be intrinsically normal, but through dilation of the ventricle and the tricuspid valve anulus it can become progressively regurgitant over time. However, more commonly, the valve is dysplastic, and this predisposes to valvular regurgitation. There is a marked association with dysplasia and displacement of the septal components of the tricuspid valve well down into the body of the right ventricle, sometimes for several centimeters, and this, in the setting of systemic pressures in the right ventricle, can create severe tricuspid valve regurgitation. We have noted double orifices in the tricuspid valve, abnormal septal clefts, and marked anular dilation. Because of difficult access and the great variability in the pathology in this valve, repair can be very difficult.18–20
Rarely, both straddling and override of the AV valves can occur, usually in association with a degree of hypoplasia of the left or right ventricle.21–23
Other Associated Cardiac Anomalies
Abnormalities of the aortic arch, including interruption of the aorta and coarctation, can occur.
Coronary Arteries
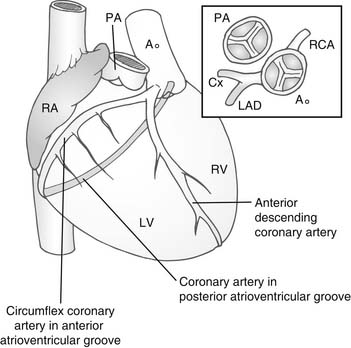
Usually, the anterior sinus of the aortic valve gives rise to what would normally be the right coronary artery (Fig. 126-2). This divides to pass down the line of the interventricular septum as the anterior descending coronary artery. The other circumflex branch passes in the AV groove between the right atrium and the left ventricle. The posterior sinus of the aortic valve gives rise to the morphologically right coronary artery passing in the AV groove between the left atrium and the right ventricle. Almost universally, the coronary artery ostia face the corresponding sinuses of the pulmonary valve. This is an important consideration in the arterial switch procedure.
Conduction System
The abnormalities of the conduction system in congenitally corrected transposition have been well described.24–27 Because of malalignment of the AV septum, there is an anterior and a superior AV node in the atrial septum adjacent to the mitral and pulmonary valve. This gives rise to a penetrating bundle that passes around the free wall of the left ventricle in a subendocardial position anterior to the pulmonary valve. The bundle then sweeps down onto the ventricular septum in the morphologic left ventricle, supplying a left bundle branch over the septal surface of the left ventricle and a penetrating right branch into the morphologic right ventricle. The sinus node is in a normal position adjacent to the entrance of the superior vena cava into the right atrium (Fig. 126-3).
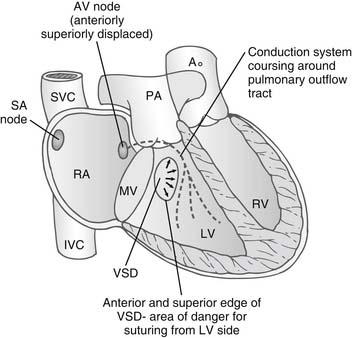
In the rarer form of situs inversus, the normally positioned AV node that is in the triangle of Koch superior to the coronary sinus persists and gives a penetrating branch to the ventricular septum but inferior and posterior to the pulmonary valve. These pathways are important because in situs solitus, when there is a VSD in the perimembranous region, the bundle passes superior to the VSD around the pulmonary outflow tract and then down on the leftward or anterior margin of the VSD. In the rarer forms of situs inversus, the bundle passes on the inferior margin of the VSD on the morphologic LV surface. Occasionally, both posterior and anterior AV nodes exist, and a loop of conduction tissue can encircle the VSD when it is present. Sutures to close the VSD are ideally placed from the morphologic RV surface of the VSD to avoid damaging the conduction system.28 The longer course of the conduction system to the ventricles associated with the malalignment of the atrioventricular septum has been given as a reason for the development of complete heart block.
CLINICAL PRESENTATION OF CONGENITALLY CORRECTED TRANSPOSITION OF THE GREAT ARTERIES
A patient’s clinical presentation depends on the presence or absence of associated cardiac anomalies. Without such anomalies, patients may be entirely free of symptoms well into late adult life. However, this is an unusual, if not rare, situation. More commonly, even in the absence of associated malformations, a degree of congestive cardiac failure develops over the lifetime of the patient, usually aggravated by the development of progressive tricuspid regurgitation.7
A small percentage of patients present in early infancy with severe congestive cardiac failure, sometimes associated with pulmonary hypertension. This is usually associated with marked tricuspid regurgitation secondary to dysplasia of the tricuspid valve, and it is often associated with a VSD that may aggravate the heart failure. Early surgical management is mandatory in these critically ill infants.18
PROBLEM OF THE TRICUSPID VALVE AND THE MORPHOLOGIC RIGHT VENTRICLE IN THE SYSTEMIC CIRCULATION
The main concern highlighted in many papers8–11,19,29–31 has been the unpredictable way the morphologic right ventricle in association with tricuspid valve regurgitation can fail when it remains as the systemic ventricle. The development of RV failure can occur primarily without previous surgical intervention and is usually associated with dysplasia of the tricuspid valve, causing tricuspid regurgitation. RV failure may also occur secondarily after conventional repair of associated cardiac defects such as VSD closure or the relief of pulmonary stenosis or atresia when the right ventricle remains in the systemic circulation.
The deterioration in the RV function associated with tricuspid regurgitation may also develop insidiously over many years without associated cardiac anomalies. There is then gradual deterioration of RV function with worsening of tricuspid regurgitation in association with the volume loading of the right ventricle. These changes may commence in childhood or even infancy and may progress at a variable rate.19,29 When associated with dysplasia or Ebsteinoid displacement of the tricuspid valve, deterioration may be rapid. Secondary deterioration of ventricular function and development of tricuspid regurgitation are recognized to occur in an unpredictable way after conventional repair of associated cardiac anomalies. Thus, VSD closure with pulmonary artery debanding may be successful with the right ventricle in the systemic circulation, although, over time, RV failure with tricuspid regurgitation can follow such a repair. In the situation of pulmonary atresia with VSD, closure of the VSD and placement of a valved conduit from the morphologic left ventricle to the pulmonary arteries may later be associated with the development of tricuspid regurgitation and RV failure.
The development of morphologic RV systemic failure without previous surgical intervention is almost always associated with dysplasia of the tricuspid valve. The suggested mechanism is that dilation of the right ventricle, in association with its volume loading resulting from tricuspid regurgitation, displaces the ventricular septum into the morphologic left ventricle. The displacement of the septal components of the tricuspid valve prevents coaptation of leaflet tissue, thus accentuating the regurgitation. This vicious cycle of tricuspid valve dilation and ventricular volume overload with increasing regurgitation continues.19 In secondary morphologic RV failure following conventional closure of VSD or relief of pulmonary valve stenosis, the fall in the morphologic LV pressure and realignment of the ventricular septum to the pulmonary ventricle can have the same effect by creating tricuspid regurgitation. That can then be aggravated by volume overloading of the morphologic right ventricle. This is thought to be the mechanism of the development of tricuspid regurgitation and RV failure, which is supported by the observation that, in placing the pulmonary artery band to train the morphologic LV, the tricuspid regurgitation can be acutely reduced by realignment of the ventricular septum.18,19 If this is the mechanism of morphologic RV failure, then tricuspid regurgitation and RV failure can be expected to occur after conventional repair.
It had been suggested that the morphologic left ventricle could be restored to the systemic circulation by redirecting the systemic and pulmonary venous returns—via a Senning or Mustard procedure—and then performing an arterial switch if the pulmonary valve and LV outflow tract were unobstructed, or a Rastelli-type procedure with rerouting of the VSD to the aorta when the VSD was suitably placed beneath the aortic valve.28 Several groups reported successful double-switch and Rastelli atrial switch procedures, showing that the morphologic left ventricle could be restored to the systemic circulation.12–1532 The underlying theme in these reports was that the morphologic left ventricle restored to the systemic circulation could be a better long-term systemic ventricle and would have the added benefit of preventing the development of RV failure. In the double-switch procedure, besides restoring the morphologic ventricle and mitral valve to the systemic circulation, the morphologic right ventricle becomes the pulmonary ventricle, works at lower pressures, and is decompressed. The amount of tricuspid regurgitation can be immediately reduced because of the lower working pressures of the right ventricle with minimal need for any repair or replacement of the tricuspid valve. Maintenance of the alignment of the ventricular septum or its displacement into the RV cavity would aid in maintaining competency of the tricuspid valve by preventing displacement of the septal leaflet of the tricuspid valve. In essence, the morphologic right ventricle is now working as a pulmonary ventricle at low pulmonary artery pressures and can function satisfactorily in that setting.
Although accentuating the inter-relationship between tricuspid valve regurgitation, right ventricular volume overloading, and right ventricular failure, several papers have highlighted the possible problem of right ventricular myocardial ischemia as a cause of right ventricular failure. The right ventricle, working at systemic pressures with resultant hypertrophy of the myocardium, may not have sufficient coronary vascular supply. This results in ischemia of the right ventricular myocardium, which, together with volume loading secondary to tricuspid valve regurgitation, may be the determining factor in RV failure in this condition.33–35
For two reasons, the double-switch type of operation has become popular over the past several years. First, at intermediate follow-up, it seems that restoration of the morphologic left ventricle to the systemic circulation with the right morphologic right ventricle working under lower pulmonary pressures prevents the development of RV failure and significant tricuspid regurgitation. The morphologic left ventricle seems to be holding up well as the systemic ventricle. In the second situation, when RV dysfunction exists with associated tricuspid regurgitation, the recruitment of the morphologic left ventricle to the systemic circulation and the delegation of the morphologic right ventricle to the pulmonary circulation is appealing, as it reduces the work that the right ventricle has to do, and the literature supports markedly diminished tricuspid valve regurgitation.36 A double-switch operation is possible when the morphologic LV pressures have been maintained at systemic levels (e.g., in a VSD pulmonary artery banded situation or when there is pulmonary atresia and VSD). However, when the morphologic left ventricle has been working as a pulmonary ventricle at low pulmonary artery pressures, it is not possible to perform a primary double-switch procedure expecting that the left ventricle will support the systemic circulation. The left ventricle needs some preparation to cope with the systemic workload. To create a ventricle strong enough to support the systemic circulation, many centers first place a pulmonary artery band to retrain the left ventricle, and later perform an arterial switch. This circumstance arises with patients who have congenitally corrected transposition without associated anomalies in whom over the first few years of life tricuspid regurgitation and a degree of RV failure has occurred in the setting of low pulmonary artery pressures. Then it is possible to retrain the left ventricle and perform a double-switch procedure once the ventricle has been retrained. It can take 6 months to 1 year, or longer, to accomplish this.
Training of the Left Ventricle
Training of the left ventricle has become an important aspect in congenital heart management since Mee’s group37 and others showed that it was possible to retrain the left ventricle in infants with dextrotransposition after a failing atrial repair to restore the left ventricle to the systemic circulation. This concept has been applied to patients with congenitally corrected transposition. The method used for training is similar in the two groups. A pulmonary artery band is placed around the main pulmonary artery acutely, to raise the morphologic LV pressure to approximately 75% to 80% of the systemic pressure. Inotrope support may be necessary, and band adjustment to loosen or tighten the band may be needed over the first few days or weeks after the initial procedure. It may take 6 to 18 months for the morphologic left ventricle to be adequately trained so that systolic pressures are at systemic levels and ventricular function is good, with an adequate thickness of the LV free wall. Training of the morphologic left ventricle is generally more successful in younger patients, and the upper age limit at which success can be expected is probably 15 or 16 years.38
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


