CHAPTER 35 Surgery for Congenital Lesions of the Esophagus
EMBRYOLOGY
At about the 18th or 19th day of fetal life, the notochord, the anlage of the vertebral column, starts to form, first in close association with endodermal cells, then separating from them. The foregut develops from the endodermal cells as they are separating from the notochord. At about 3 weeks of embryonic development, the tracheal primordium appears as a ventral diverticulum in the cephalad portion of the foregut. During the next few weeks, growth and elongation of the diverticulum and the foregut along the tracheoesophageal groove contribute to the separation of the esophagus and the trachea, which is complete by about 5 to 6 weeks of fetal life. During the seventh and eighth weeks, the esophageal epithelium proliferates and fills the lumen almost completely. Vacuoles appear in the lumen and eventually coalesce to recanalize it by the tenth week.1
A doxorubicin (Adriamycin)–induced murine model of EA has been described.2 With use of this model, investigators are studying the role that patterning genes and proteins like Sonic hedgehog (Shh) might play in the morphogenesis of EA.3,4 In the same rat model as well as in neonates with EA/TEF, Spilde and coworkers have studied the molecular expression of foregut patterning genes to shed a light on the origin of TEF. The distal esophagus seems to arise as a diverticulum of the trachea, which elongates and joins the stomach, rather than from the foregut itself.2,5,6 They speculate that this might explain the well-known poor motility of the reconstructed esophagus in patients with TEF.
ESOPHAGEAL ATRESIA
Historical Aspects
The historical background relevant to EA is thoroughly reviewed by Harmon and Coran.7 Durston in 1670 and Gibson in 1697 described the first cases of EA. It took about 250 years before the first reported cases of survivors by Leven and Ladd independently in 1939. Both were able to achieve success by performing a series of operations including gastrostomy, ligation of the fistula, marsupialization of the upper pouch, and final reconstruction with an antethoracic skin tube. The early attempts at primary repair were all unsuccessful. It was not until 1941 that Haight reported the first survivor of a primary repair. In the decade that followed, it became evident that the mortality was very high in infants of lower birth weights, in those with severe associated anomalies, and in those critically ill from aspiration pneumonia.8–10 There followed a shift toward staging of the operation for sick infants, with a gastrostomy followed by division of the TEF and the esophageal reconstruction performed as a third stage.8–10 In 1962, Waterston proposed a classification based on birth weight, presence of pneumonia, and associated anomalies.9 The 1970s and 1980s witnessed major advances in respiratory, neonatal, anesthetic, and surgical care as well as the introduction of more effective antibiotics. These advances included endotracheal intubation, which made it easier to prevent aspiration from the esophageal pouch and to deal with its sequelae. As a result, multiple groups started to recommend either direct primary anastomosis (anastomosis shortly after birth) or delayed primary anastomosis (anastomosis delayed for the treatment of other life-threatening anomalies or stabilization of the patient) regardless of the patient’s weight but based on physiologic criteria.11–19 The end of the 20th century ushered in the application of thoracoscopy to the repair of EA/TEF and other congenital anomalies of the esophagus.20–27
Epidemiology
The average rate of EA is reported to be about 2.4 per 10,000 births.28 No significant sex predilection is described. Other congenital anomalies occur in patients with EA frequently, ranging from 30% to 76%.29–33 This might be due to the fact that the malformation in EA occurs early in the first trimester when there is active organogenesis. As a result, the developmental cause of EA/TEF might also affect other organ systems at the same time. The number of associated anomalies occurring in each patient increases with decreasing birth weight.29,34,35 With the improvement in anesthetic, respiratory, and neonatal techniques during the last few decades, the associated anomalies are now the major contributor to mortality in patients with EA.31 The most common associated anomaly is congenital heart disease, present in some form in about a fifth of the patients.29,33–37 About 20% of patients have some combination of the constellation of anomalies referred to as VATER or VACTERL association: vertebral, anorectal, cardiac, tracheoesophageal, renal or radial, and limb anomalies.36,37
Infants born with EA often have low birth weight and are premature.35,38 In one study, 90% of the patients with EA were below the 50th percentile for gestational age, and 40% were below the 10th percentile or small for gestational age.39 The growth retardation might be secondary to decreased absorption of the amniotic fluid protein or a mechanical factor.39 Severe intrauterine growth retardation increases the mortality rate of the neonate who is small for gestational age by 5 to 20 times that of neonates who are appropriate for gestational age.40
Anatomy
There are five types of EA with or without TEF. Different classification schemes have numbered them differently, so it is preferred to describe the actual anomaly rather than assign it a number or a letter (Fig. 35-1): EA with distal TEF, EA without TEF, EA with proximal TEF, EA with proximal and distal fistula, and isolated TEF (H-type TEF). The distribution of the different types in large series has been relatively uniform across different decades and countries, with the most common being EA with distal TEF (Table 35-1).11,29,41–43 The fistula is usually small and most of the time arises from the midline of the membranous portion of the trachea just above the bifurcation, but there are significant variations.
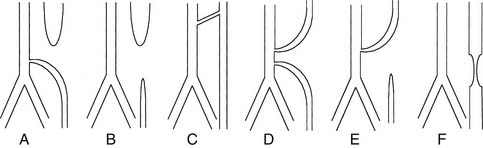
Table 35–1 Distribution of Types of Esophageal Atresia
| Type of Anomaly | Number | Percentage |
|---|---|---|
| EA with distal TEF | 1024 | 87.1 |
| EA | 82 | 7.0 |
| H-type TEF | 37 | 3.1 |
| EA with proximal TEF | 11 | 0.9 |
| EA with double TEF | 22 | 1.9 |
EA, esophageal atresia; TEF, tracheoesophageal fistula.
Presentation
A significant number of cases of EA are now suspected on prenatal ultrasonography; polyhydramnios, absent or small stomach bubble, and visualization of an esophageal pouch in the neck are the most prominent features.44,45 Suspecting the diagnosis prenatally is invaluable in preparing the family. Prenatal counseling with a pediatric surgeon and a neonatologist and planning for appropriate delivery arrangements are extremely helpful. Postnatally, EA is diagnosed in most patients in the first few hours after birth. Choking with feeding, regurgitation of saliva and feeds, and respiratory distress from aspiration of saliva or gastric contents through the TEF are the most common signs and symptoms. Inability to pass a feeding tube confirms the diagnosis.
Isolated TEF (H-type TEF) may not be diagnosed until later in life. Recurrent episodes of aspiration pneumonia and choking and coughing with feedings should raise the suspicion. Contrast esophagography and rigid bronchoscopy are complementary in making the diagnosis.46
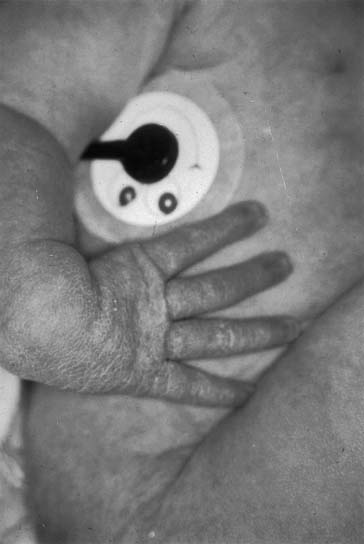
Patients with isolated EA often have a scaphoid abdomen because of the absence of gas in the intestines. If EA is suspected, one should always look for other physical signs of the VATER association: anorectal malformations, limb anomalies, and vertebral defects (Fig. 35-2).
Workup
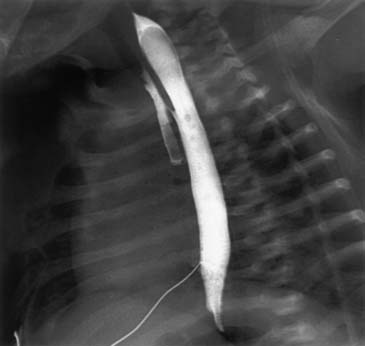
A chest radiograph showing a curved catheter in the proximal esophageal pouch is often all that is required to make the diagnosis (Fig. 35-3). In patients with isolated EA, the radiograph reveals absence of intestinal air (Fig. 35-4). If there is still doubt, a small amount of air injected into the pouch accentuates it on a plain radiograph and confirms EA (Fig. 35-5). The use of barium to look for an upper pouch fistula should be discouraged because it may potentially lead to aspiration. Upper pouch fistulas are rare and are usually found at the time of repair by either bronchoscopy or a careful dissection of the proximal pouch. Esophagography performed through a catheter being pulled up along the esophagus while the patient is prone is invaluable in making the diagnosis of H-type fistula (Fig. 35-6).
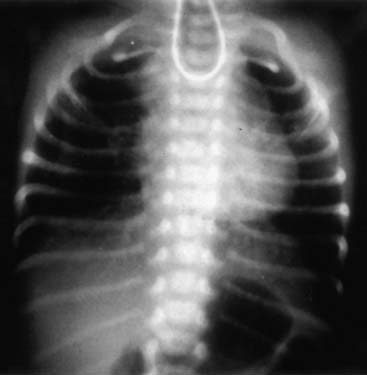
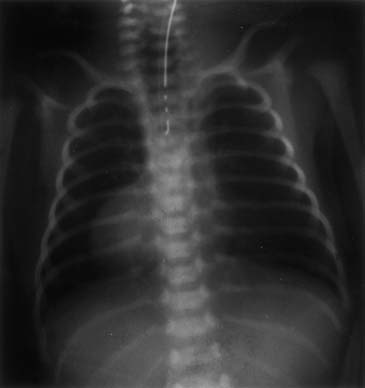
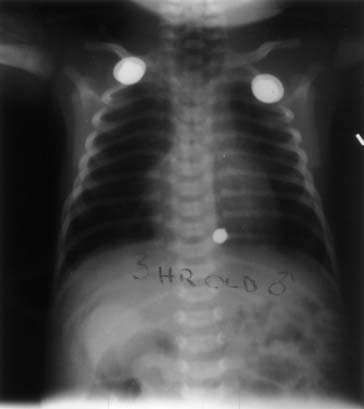
Figure 35–5 Air-contrast esophagogram in patient with esophageal atresia showing the distended proximal pouch.
(Courtesy of Dr. C. Leftridge, Georgetown University Medical Center.)
Initial Management
Positive-pressure ventilation should be avoided in a patient with a TEF if at all possible to minimize shunting through the TEF and abdominal distention. Because the majority of the fistulas are located just proximal to the carina, the tip of the tube should be kept high in the trachea to prevent the tip of the tube from getting lodged in the TEF. The TEF is sometimes significant enough that adequate ventilation cannot be maintained, especially in the face of respiratory distress syndrome of the premature with its attendant high intraparenchymal pressures. In that case, emergent ligation of the TEF might be warranted.47 A more difficult way to control the TEF emergently is obliteration of the TEF with a Fogarty balloon introduced by bronchoscopy.48 A hazardous situation can occur when a patient with significant steal through a TEF also has a very high intestinal obstruction, such as duodenal atresia. The massive gastric distention exacerbates the respiratory compromise and could lead to perforation of the stomach. Emergent gastric decompression has to be performed, sometimes at the bedside with a needle.
The timing of repair of EA is of particular interest. The key issues affecting the timing include severity of the infant’s condition, associated abnormalities, and birth weight. The Spitz classification acknowledges low birth weight (<1500 g) and major congenital heart disease as two major risk factors associated with EA.49 This system stratifies patients into groups. Survival was estimated at 97% for group I (weight >1500 g with no major congenital cardiac defect), 59% for group II (birth weight <1500 g or major congenital cardiac defect), and 22% for group III (birth weight <1500 g and major congenital cardiac defect). In 2006, Lopez and colleagues50 examined the same risk factors in 188 neonates and found survival results comparable to those of Spitz, thus validating the Spitz classification.
Operative Principles
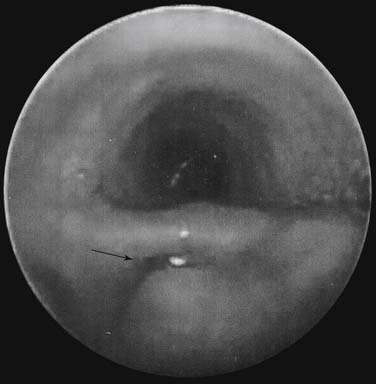
Rigid bronchoscopy is helpful at the beginning of the procedure to identify the exact location of the TEF, to recognize rare variants like double fistulas or H-type fistula and laryngotracheoesophageal clefts, to identify tracheomalacia, and to help in placement of the endotracheal tube to avoid dislodgement into the TEF (Fig. 35-7).51–53
Primary repair of EA with division of the TEF and end-to-end anastomosis is the ideal goal (Fig. 35-8). The standard approach is a right posterolateral thoracotomy. If the patient has a right-sided aortic arch, it might be easier to approach the esophagus from the left thorax. Having the patient tilted forward in the nearly prone position facilitates access to the posterior mediastinum. To minimize some of the complications reported with thoracotomy in neonates, namely, winged scapula and scoliosis, an axillary skin crease thoracotomy has been reported by Bianchi54 and used with good results.55 Traditionally, an extrapleural approach has been advocated to decrease the risk of empyema if an esophageal leak occurs. With the introduction of more powerful antibiotics in the 1970s and 1980s, the importance of a retropleural approach with the potential increase in operative time has been questioned.41,56
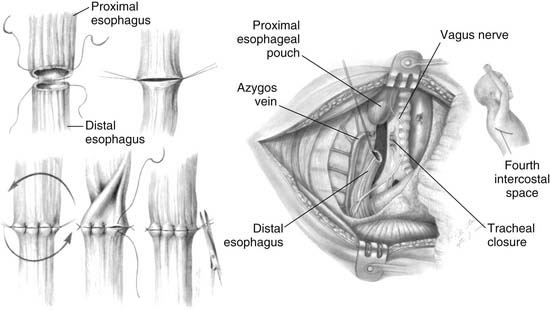
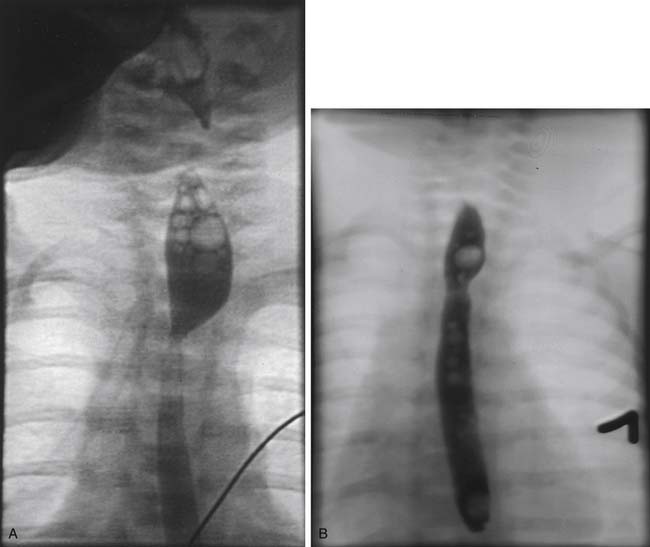
Gentle pressure by the anesthesiologist on the pouch catheter helps identify the upper pouch, which is usually high in the thoracic inlet. A transmural suture placed through the fistula and incorporating the catheter makes the manipulation of the upper esophagus less traumatic. The upper pouch and the trachea are intimately juxtaposed, often sharing a common wall. The dissection between the esophageal pouch and the trachea is delicate. Extreme caution should be applied to avoid injury to both vagus and recurrent laryngeal nerves. The pouch is mobilized as high as possible to minimize the tension of the anastomosis. The blood supply of the upper esophagus is intramural, allowing minimal ischemia even after extensive mobilization. In contradistinction, the lower esophagus is supplied by segmental branches from the aorta; therefore, its mobilization should be minimized to prevent ischemia. The ends of the esophagus are trimmed, and an end-to-end anastomosis is built in a single-layer fashion with a fine monofilament absorbable suture. The knots are tied extraluminally if possible. It is crucial to identify the mucosa of both the upper and lower esophagus and to incorporate it in the sutures. If there is significant tension, it is helpful to leave the sutures untied and to approximate all at the same time as the knots are tied to take some of the tension off. The esophageal and tracheal suture lines must be separated to avoid fistula formation. This is usually accomplished by the interposition of a pleural flap, but a pericardial flap is sometimes required. The routine use of gastrostomy and transanastomotic feeding tubes remains controversial.7 Before the anastomosis is constructed, congenital esophageal stenosis needs to be ruled out by passing a tube through the distal esophagus into the stomach.57 At the completion of the procedure, a small chest tube is placed and secured to the endothoracic fascia away from the anastomosis. At about 5 or 7 days postoperatively, a contrast study is obtained to assess the anastomosis. The disparity in size between the distended proximal pouch and the small distal esophagus gives the appearance of a narrowing, but usually prompt emptying of contrast material attests to the wide patency of the anastomosis (Fig. 35-9A). With time, the size discrepancy becomes less pronounced (Fig. 35-9B). If there is no leak, the chest tube is removed and feedings are started. Because of the frequent occurrence of gastroesophageal reflux and the deleterious effects acid can have on a fresh anastomosis, serious consideration should be given to keeping the patient on acid suppressive and promotility drugs until the anastomosis is well healed.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


