CHAPTER 124 Surgery for Congenital Anomalies of the Coronary Arteries
In most individuals, each coronary artery ostia is located centrally in the appropriate sinus of Valsalva. In some patients, the ostium may be eccentrically located, and in others a coronary artery may arise close to a valve commissure. One or both coronary ostia may arise from the tubular aorta above the sinotubular junction, which is usually a benign finding. However, this anomaly becomes significant if an aortotomy is necessary for aortic valve replacement or for another indication; if not recognized, the coronary artery could be transected. Another anomaly is when both coronary arteries arise from the same aortic sinus with either a single ostium or two separate ostia (Box 124-1). This is generally not clinically significant if the aberrant vessel courses posterior to the aorta or anterior to the pulmonary artery, but it becomes important if either the aberrant LMCA or RCA courses between the two great vessels, as this may lead to myocardial ischemia and sudden death.
Box 124–1 Origin of Both Coronary Arteries from One Sinus
Reprinted with permission from Gaynor JW. Coronary artery anomalies in children. In: Kaiser LR, Kron IL, Spray TL, editors. Mastery of cardiothoracic surgery. Philadelphia: Lippincott-Raven; 1998, p. 881.
ANOMALOUS ORIGIN OF A CORONARY ARTERY FROM THE PULMONARY ARTERY
Anomalous origin of a coronary artery from the pulmonary artery is a rare congenital anomaly that is almost always fatal if not diagnosed and treated.1,2 Although anomalous origin of the LMCA from the pulmonary artery (ALCAPA) is the most common, other coronary arteries may rarely also arise from the pulmonary artery. The RCA arising from the pulmonary artery is found approximately 10 times less frequently than ALCAPA and is also associated with ischemia and sudden cardiac death. The LAD, the circumflex, or both the left and right coronary arteries may arise anomalously from the pulmonary artery; these variants are extremely rare and are almost uniformly fatal.
ALCAPA is the most important congenital coronary artery anomaly in this class. It is a very rare lesion with an incidence ranging from 1 in 30,000 to 1 in 300,000 people. It is the most common cause of myocardial infarction in childhood, and, if left untreated, there is a 90% mortality rate by age 1 year. ALCAPA is also known as the Bland-White-Garland syndrome, after Bland and colleagues reported on both clinical and autopsy findings in an infant with this anomaly in 1933.3
Anatomy
In ALCAPA, the LMCA usually arises from the main pulmonary artery (MPA); occasionally, it arises from the right pulmonary artery. It usually originates from the rightward aspect of the posterior (facing) sinus of the MPA (Figs. 124-1 and 124-2), but it may also originate from the leftward aspect of the posterior (facing) and rarely from the anterior (nonfacing) sinus of the MPA (see Fig. 124-2). An anomalous RCA most commonly originates from the anterior portion of the pulmonary artery.
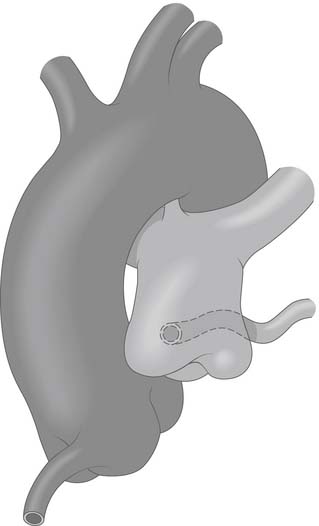
Figure 124–1 The aorta and pulmonary artery showing anomalous origin of the left main coronary artery from the posterior-facing sinus of the pulmonary artery with the anomalous artery coursing behind the pulmonary artery www.lww.com.
(Reprinted with permission from Gaynor JW. Coronary artery anomalies in children. In: Kaiser LR, Kron IL, Spray TL, editors. Mastery of cardiothoracic surgery. Philadelphia: Lippincott-Raven; 1998, p. 881.)
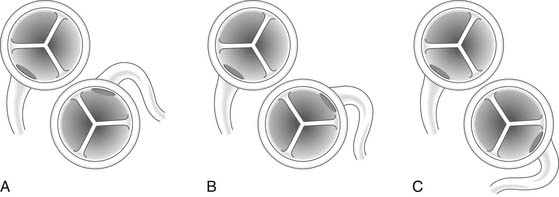
Figure 124–2 A, Anomalous origin of the left main coronary artery from the rightward aspect of the posterior-facing sinus of the pulmonary artery. B, Anomalous origin of the left main coronary artery from the leftward aspect of the posterior-facing sinus. C, Anomalous origin of the left main coronary artery from the nonfacing sinus of the pulmonary artery www.lww.com.
(Reprinted with permission from Gaynor JW. Coronary artery anomalies in children. In: Kaiser LR, Kron IL, Spray TL, editors. Mastery of cardiothoracic surgery. Philadelphia: Lippincott-Raven; 1998, p. 882.)
Pathophysiology
Children with ALCAPA usually develop symptoms after ductal closure and the subsequent fall in pulmonary vascular resistance. During fetal life, the pressures of the systemic and pulmonary circulations are similar, and myocardial perfusion remains intact because the pulmonary arterial pressure is systemic. After the child is born but before ductal closure, the pulmonary artery pressure remains elevated, and perfusion of the anomalous coronary artery is maintained. Because of this, children are rarely diagnosed with ALCAPA in the first few days of life. The clinical course after ductal closure is largely determined by the presence or absence of collaterals from the RCA to the left coronary system. If there is inadequate collateral circulation, myocardial ischemia and ventricular dysfunction result from insufficient myocardial perfusion.4 This is because the pulmonary artery pressure is now well below the systemic pressure, and the left ventricle is thus perfused with desaturated blood at a low pressure. However, if there is elevated pulmonary artery pressure because of the presence of a PDA or a VSD, left ventricular perfusion pressure may be adequate and ischemia may not occur. If this anomaly is unknown before the closure of these defects, it will become apparent shortly after closure with the subsequent drop in pulmonary arterial pressure—usually with fatal outcome.
If collaterals are adequate, perfusion of the left coronary system is maintained. As the pulmonary vascular resistance falls, however, a left-to-right shunt develops from the RCA to the pulmonary artery. There is progressive dilation of the RCA and left coronary artery systems, with reversal of flow in the left coronary leading to a pulmonary–coronary steal. Although the shunt is relatively small compared with overall cardiac output, it is significant with regard to coronary blood flow. Children with significant collaterals may survive past infancy; however, there is usually progressive left ventricular dysfunction.5 In a small percentage, the collateral vessels are enough to maintain adequate myocardial perfusion at rest and sometimes even during exertion, and these patients may not present until adulthood.6 In most children, however, severe mitral regurgitation is present secondary to papillary muscle dysfunction and ventricular dilation.
Clinical Presentation
Infants frequently present between 4 and 6 weeks of age when the pulmonary vascular resistance has dropped.7 However, infants may not present until closer to 2 to 3 months of age, when symptoms have become more severe. Infants usually present with signs and symptoms of congestive heart failure, including sweating and discomfort with feeding, tachypnea, poor weight gain, and pallor. The discomfort with feeding most likely represents myocardial ischemia. Children who do not present as infants may be diagnosed at several months of age as a result of a loud murmur of mitral regurgitation. Older children, adolescents, and adults may remain asymptomatic, whereas others may come to clinical attention because of exertional chest pain, pre-syncope, or syncope. There have been reports of sudden death with exercise in these older patients. The symptoms associated with anomalous origin of the RCA from the pulmonary artery are less severe, but myocardial ischemia and death can still occur.
Diagnostic Imaging
Cardiac catheterization with angiography remains the gold standard in the diagnosis of ALCAPA. In infants presenting with ALCAPA, cardiac catheterization demonstrates elevated filling and pulmonary arterial pressures and a low cardiac output. In older patients who are asymptomatic, it may show only mildly elevated pulmonary arterial pressures but normal filling pressures and cardiac output. A small left-to-right shunt may be present. An aortogram will demonstrate a single, dilated RCA arising normally from the aorta. If significant collaterals are present, aortic root angiography will show the collaterals providing late, retrograde filling of the left coronary artery with a blush of contrast subsequently filling the MPA. A step-up in oxygen saturation may be noted in the MPA when there is a large left-to-right shunt from the collaterals. If doubt remains regarding the diagnosis, a main pulmonary arteriogram with distal balloon occlusion should demonstrate the anomalous left coronary artery.8
Magnetic resonance imaging (MRI) has emerged as a useful noninvasive diagnostic tool for delineating congenital coronary anomalies.9,10 Although there have been case reports of using this method in diagnosing ALCAPA, no case series with this anomaly have been reported. However, studies have shown that magnetic resonance angiography has a similar sensitivity and specificity when compared with coronary angiography and may be especially helpful in delineating the proximal course of anomalous coronary arteries. Computed tomography (CT) has been used extensively for coronary artery delineation in adults. Advantages of this technique include rapid acquisition time and high resolution, but the disadvantages include radiation exposure and the need for a slower heart rate with ECG gating, precluding its use in infants.
Surgical Management
Indications for surgery
Surgical repair is indicated in all patients with ALCAPA. In infants who present in congestive heart failure, surgery should occur within the first few days of diagnosis because risk of continuing myocardial ischemia and death is very high.11 In older patients who present without symptoms, surgery is necessary but can be performed electively. In an infant who presents in severe heart failure secondary to myocardial infarction, surgery will probably need to be delayed for at least 24 hours to stabilize the patient using mechanical ventilation, inotropic support, and vasodilators, when necessary. Del Nido and colleagues reported that even in the sickest infants, a two-vessel surgical repair is possible if left ventricular assist devices (LVAD) are used postoperatively.12 Others may need extracorporeal membrane oxygenation (ECMO). Because infants requiring surgery are often quite ill, it is important that centers performing ALCAPA surgery have these options available, or else the child should be transferred to a hospital with these capabilities.
The goal of surgery is restoration of a two-coronary system; therefore, simple ligation of the anomalous coronary should not be performed.13 Severe left ventricular dysfunction and mitral insufficiency are not contraindications to revascularization in infants, because significant recovery of function usually occurs. Left ventricular aneurysmectomy and mitral valve repair or replacement are rarely indicated at the time of initial procedure even if severe mitral regurgitation is present, because the severity of mitral regurgitation almost always decreases after revascularization.
Surgical Techniques
Bypass grafts were accomplished using the left subclavian artery, the internal mammary artery (IMA), and the saphenous vein. Meyer and colleagues reported the first successful left subclavian artery–to–left coronary artery bypass in 1968.14 The results of bypass grafting, especially with saphenous vein grafts, have been disappointing. Takeuchi and associates were the first to describe the creation of an aortopulmonary window and intrapulmonary artery baffle using a pulmonary artery flap to direct blood flow from the aorta to the anomalous coronary artery.15 Finally, direct reimplantation of the anomalous coronary into the aorta has become the procedure of choice in many institutions, as experience with the arterial switch operation for transposition of the great vessels has increased.16,17
Coronary Artery Bypass Grafting
Coronary artery bypass grafting is rarely used in patients with ALCAPA. In the current era, it is usually used to create a dual coronary artery system after previous ligation or because of stenosis or occlusion after a previous attempt at repair. The IMA is the conduit of choice and can be successfully used even in neonates and infants, and there is some evidence for growth of the IMA after bypass grafting.18–20 The saphenous vein should not be used unless it is the only conduit available, because of the risk of occlusion and poor long-term results.21 Similarly, left subclavian–to–left coronary artery anastomosis is infrequently used because of the risk of anastomotic stenosis or occlusion.22,23
Direct Reimplantation
In most patients with ALCAPA, direct reimplantation of the anomalous coronary artery onto the aorta can be performed (Fig. 124-3).24,25 When the anomalous coronary ostium is in the posterior-facing sinus, the procedure is fairly straightforward. Direct implantation is possible even if the ostium is located in the nonfacing sinus by excising a large button of pulmonary artery to extend the coronary artery.
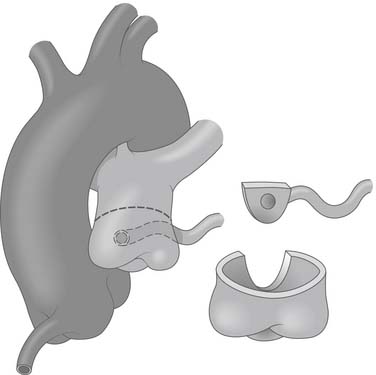
Figure 124–3 After institution of cardiopulmonary bypass and induction of cardioplegia, the pulmonary artery is transected above the sinotubular junction, and the anomalous coronary ostium is excised with a generous button of pulmonary artery wall www.lww.com.
(Reprinted with permission from Gaynor JW. Coronary artery anomalies in children. In: Kaiser LR, Kron IL, Spray TL, editors. Mastery of cardiothoracic surgery. Philadelphia: Lippincott-Raven; 1998, p. 883.)
The aorta and both pulmonary arteries are fully mobilized. To improve mobility of the pulmonary artery, the ductus (or ligamentum) arteriosus is ligated. Tourniquets are placed around both the right and left branch pulmonary arteries to occlude the branch pulmonary arteries and prevent runoff of cardioplegic solution into the lungs. Another option to prevent runoff is to compress the origin of the coronary artery from the pulmonary artery during administration of cardioplegic solution. A cannula is inserted in the ascending aorta for administration of cardioplegia solution, the aorta is cross-clamped, and cold cardioplegia is administered via the aortic root. If circulatory arrest is used, the head vessels are then occluded with tourniquets, the circulation is arrested, venous blood is drained into the reservoir, and the cannulas are removed. After adequate arrest, the pulmonary artery is transversely opened just above the sinotubular junction (see Fig. 124-3). The anomalous coronary orifice is identified. The pulmonary artery is divided and the coronary ostium is excised from the pulmonary artery with a generous button of arterial wall, as in the procedure used for the arterial switch operation. The segment of the pulmonary wall that is excised extends the proximal end of the coronary artery, thus allowing the aortic anastomosis to be accomplished without tension. To excise the coronary button, the pulmonary commissure may need to be taken down if the coronary ostium is located near a commissure. An alternative technique that can be used if the coronary arises anteriorly from the pulmonary artery or from a branch pulmonary artery involves extension of the coronary artery with a tube constructed from pulmonary artery wall to allow reimplantation (Fig. 124-4). The proximal portion of the coronary artery is mobilized with cautery, using caution to avoid any small branches. As in the arterial switch operation, the aorta is then opened transversely immediately above the sinotubular junction, and the incision is carried posteriorly above the left posterior sinus (Fig. 124-5). The sinus is then incised vertically to accept the coronary button. The coronary button is carefully aligned with the aortic incision to avoid twisting or kinking. A continuous suture of 7-0 polypropylene (Prolene) is used to start the anastomosis at the most inferior aspect of the coronary button, which is attached to the most inferior aspect of the incision in the sinus. The suture line is carried to the top of the incision anteriorly and posteriorly. The aorta is closed with a continuous suture of 7-0 Prolene, which is tied to the coronary button suture as the anastomosis is completed (see Fig. 124-5). Once the aorta has been closed, cardioplegia solution is administered and the anastomotic site is inspected for adequate filling of the coronary and for hemostasis.
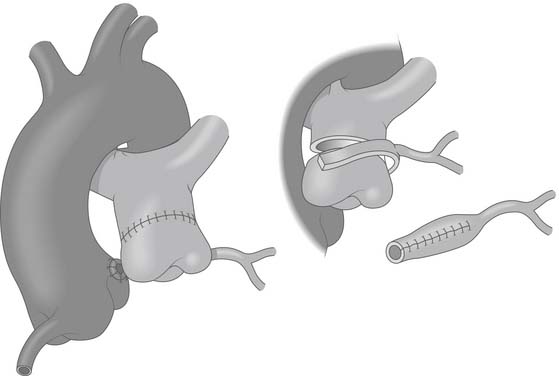
Figure 124–4 Occasionally when the coronary artery arises from the leftward or anterior aspect of the pulmonary artery, direct reimplantation may not be possible. In these situations, a tube can be constructed from a segment of pulmonary artery to lengthen the coronary and allow reimplantation on the aorta www.lww.com.
(Reprinted with permission from Gaynor JW. Coronary artery anomalies in children. In: Kaiser LR, Kron IL, Spray TL, editors. Mastery of cardiothoracic surgery. Philadelphia: Lippincott-Raven; 1998, p. 883.)
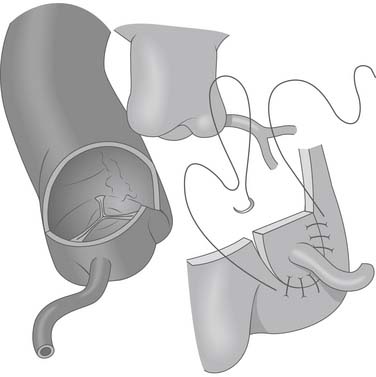
Figure 124–5 After the anomalous coronary artery is mobilized, the aorta is opened transversely above the sinotubular junction and a vertical incision is made in the left posterior sinus to accept the reimplanted coronary www.lww.com.
(Reprinted with permission from Gaynor JW. Coronary artery anomalies in children. In: Kaiser LR, Kron IL, Spray TL, editors. Mastery of cardiothoracic surgery. Philadelphia: Lippincott-Raven; 1998, p. 884.)
In the majority of cases, the pulmonary artery can be repaired primarily with a continuous suture of 7-0 Prolene (Fig. 124-6). The ductus should be divided to improve mobility of the pulmonary artery confluence, thus allowing reconstruction without tension. However, if there is tension or narrowing, the pulmonary artery should be repaired with a patch of autologous pericardium (see Fig. 124-6). If a commissure was taken down during excision of the coronary button, the pulmonary artery should be reconstructed with pericardium and the commissure resuspended.
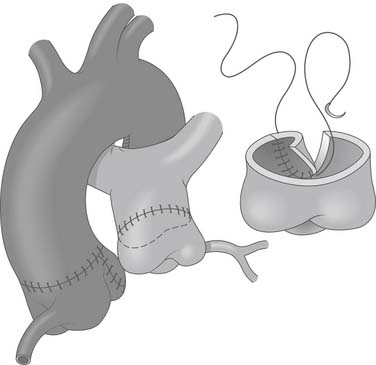
Figure 124–6 After the coronary is reimplanted, the aorta is closed primarily. The pulmonary artery may often be closed primarily. Ligation and division of the ligamentum arteriosus improves mobility of pulmonary artery. Occasionally, patch repair of the defect in the pulmonary artery with autologous pericardium may be necessary (inset) www.lww.com.
(Reprinted with permission from Gaynor JW. Coronary artery anomalies in children. In: Kaiser LR, Kron IL, Spray TL, editors. Mastery of cardiothoracic surgery. Philadelphia: Lippincott-Raven; 1998, p. 884.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


