Sudden Death Syndromes
The most common etiology of sudden cardiac death (SCD) is acute ischemia or ventricular tachycardia (VT) resulting from prior myocardial infarction. Dilated cardiomyopathy from a nonischemic cause is another important substrate for ventricular arrhythmias and sudden death. These two entities are discussed in separate chapters. The sudden death syndromes due to inherited arrhythmias or acquired electrical abnormalities are discussed in this chapter. The inherited arrhythmia syndromes are physiologically diverse disorders. They are, however, often considered as a group because of important shared clinical features:
An increased risk of malignant ventricular arrhythmias and sudden cardiac arrest (SCA)
Presentation in children, adolescents, and young adults
In most syndromes, an absence of overt structural heart disease
In practice, the cardiologist or electrophysiologist is often asked to consider all of these diagnoses in young, otherwise healthy, individuals who present with palpitations, presyncope, syncope, nonsustained ventricular arrhythmias, or cardiac arrest.
Because of the serious implications of the diagnoses and the nonspecific nature of the common presenting complaints, a disciplined, systematic
approach to the consideration of these disorders is necessary. The cornerstones of such an evaluation remain the clinical history and close evaluation of the electrocardiogram (ECG) although advanced testing including cardiac magnetic resonance imaging (MRI), genetic testing, electrophysiology (EP) testing, and microvolt T wave alternans (TWA) also have a role in selected cases.
approach to the consideration of these disorders is necessary. The cornerstones of such an evaluation remain the clinical history and close evaluation of the electrocardiogram (ECG) although advanced testing including cardiac magnetic resonance imaging (MRI), genetic testing, electrophysiology (EP) testing, and microvolt T wave alternans (TWA) also have a role in selected cases.
The list of disorders included in the inherited arrhythmia syndromes varies and it has evolved over the last 20 to 30 years, reflecting the ongoing recognition, definition, and characterization of these diseases. In this discussion, we will include six disorders in two general groups; ion channelopathies and diseases of the myocardium:
Ion channelopathies
Congenital long QT syndrome (LQTS)
Short QT syndrome
Brugada syndrome (BSM)
Catecholaminergic polymorphic VT
Myocardial diseases
Arrhythmogenic right ventricular dysplasia (ARVD)
Hypertrophic cardiomyopathy (HCM)
Less common diseases, including Rett syndrome, and Timothy syndrome, are also associated with an increased risk of SCA. Owing to their rarity and/or their association with other, more prominent, noncardiac abnormalities, these disorders are not discussed in detail here.
Long QT Syndrome
The congenital LQTS is a disorder of myocardial repolarization. It is characterized by prolongation of the QT interval, morphologic abnormalities of the T wave, and an increased risk of torsade de pointes (TDP). Traditionally, the congenital LQTS has been characterized as an autosomal dominant disorder called the Romano-Ward syndrome or an autosomal recessive disorder called the Jervell and Lange-Nielsen (JLN) syndrome. The latter syndrome has associated congenital sensorineural deafness. It is now recognized that most forms of congenital LQTS result from mutations in the genes responsible for the cardiac potassium and sodium channels. The acquired form of long QTS is often related to electrolyte depletion or drug exposure, particularly with antiarrhythmic agents that block potassium channels (see Table 13-1). It is currently
believed that many patients with acquired LQTS have a partial abnormality in the HERG potassium channel, which is clinically inapparent until exposure to a potassium channel blocking drug or severe electrolyte depletion results in marked QT prolongation.
believed that many patients with acquired LQTS have a partial abnormality in the HERG potassium channel, which is clinically inapparent until exposure to a potassium channel blocking drug or severe electrolyte depletion results in marked QT prolongation.
TABLE 13-1 Common Causes of Acquired Long QT Syndrome (LQTS)
| SSRI, selective serotonin reuptake inhibitor; AV, atrioventricular; HIV, human immunodeficiency virus. |
| Antiarrhythmic drugs: quinidine, procainamide, disopyramide, sotalol, dofetilide, amiodarone, ibutilide |
| Antihistamines (particularly in association with erythromycin): terfenadine, astemizole |
| Psychotropic drugs: thioridazine, haloperidol (Haldol), tricyclic antidepressants, methadone, risperidone, SSRIs, phenothiazines |
| Antibiotics: erythromycin, clarithromycin, azithromycin, |
| Metabolic causes: hypokalemia, hypomagnesemia (particularly, in association with diuretics), anorexia nervosa, liquid protein diets |
| Bradyarrhythmias: AV block |
| HIV protease inhibitors |
The accuracy of data on the epidemiology of the congenital LQTS is limited by both variable penetrance of the clinical phenotype and incomplete genetic characterization of the disease. Estimates of the incidence of LQTS range between 1 in 2,500 and 1 in 10,000.
Seven genes have been linked to congenital LQTS (see Table 13-2). Five encode proteins that are components of cardiac potassium channels, one the cardiac sodium channel, and one the plasma membrane protein ankyrin B. Mutations in at least one of these loci have been detected in up to 70% of patients with congenital LQTS.
In addition to LQT 1 to 7, QT prolongation and malignant ventricular arrhythmias have been identified in patients with other genetic disorders, including Rett syndrome, a neurodevelopmental disorder affecting approximately 1/20,000 females; and Timothy syndrome, a rare multisystem disorder with a total of 17 described cases.
Long QT Clinical Syndrome
The clinical presentation of congenital LQTS is most often syncope, seizures, or sudden death due to TDP. The average age of clinical presentation is 7 with one third of patients having a family history of LQTS. LQT-associated TDP can be misdiagnosed as a seizure disorder particularly in children. The circumstances
associated with the cardiac event (i.e., syncope, sudden death) are also helpful. Patients with LQT 1 often have exercise-related events. TDP associated with swimming is typical with LQT 1 and may be a cause of unexplained drowning. LQTS 2 is unique in that startling auditory stimuli can precipitate arrhythmic events (e.g., “alarm clock syncope”). LQT 3 is characterized by arrhythmias occurring at rest or during sleep.
associated with the cardiac event (i.e., syncope, sudden death) are also helpful. Patients with LQT 1 often have exercise-related events. TDP associated with swimming is typical with LQT 1 and may be a cause of unexplained drowning. LQTS 2 is unique in that startling auditory stimuli can precipitate arrhythmic events (e.g., “alarm clock syncope”). LQT 3 is characterized by arrhythmias occurring at rest or during sleep.
TABLE 13-2 Genes Linked to Congenital Long QT Syndrome (LQTS)
| Syndrome | Gene | Current | Action Potential Effect | LQTS Syndrome | Percentage of LQTS (%) |
| RW, Romano Ward; JLN, Jervell and Lange-Nielsen; HPP, Hereditary periodic paralysis. | |||||
| LQT 1 | KvLQ1 | IKs | Delayed phase 3 | RW/JLN | 42–55 |
| LQT 2 | HERG | IKr | Delayed phase 3 | RW | 35–45 |
| LQT 3 | SCN5A | Ina | Prolonged phase 2 | RW | 8–10 |
| LQT 4 | Ankyrin B | — | — | RW | — |
| LQT 5 | KCNE1 | IKs | Delayed phase 3 | RW/JLN | — |
| LQT 6 | KCNE2 | IKr | Delayed phase 3 | RW | — |
| LQT 7 | KCNJ2 | Klr2.1 | Delayed phase 3 | HPP | — |
Long QT Syndrome Diagnosis
A variety of tests and algorithms have been proposed for diagnosing LQTS including the Schwartz score. This score comprises the most clinically important factors associated with risk of clinical events with LQTS and assigns points for the degree of QT prolongation (e.g., ≥480 msec, family history, personal history of syncope, and T-wave morphology).
In genotypically confirmed LQTS, longer QT intervals, particularly those >500 msec, are associated with a higher frequency of cardiac events. Importantly, 6% of patients with LQTS will have cardiac events with a normal QT interval. The T-wave morphology can help predict the specific type of LQT. Specifically, a broad T wave suggests LQT 1, a notched T wave suggests LQT 2, and a long isoelectric ST segment and T wave, LQT 3 (see Fig. 13-1).
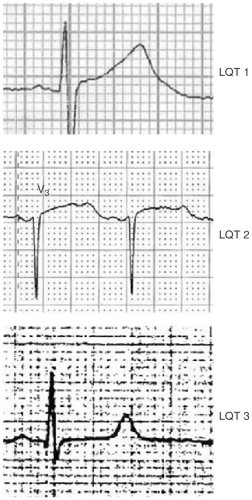 FIGURE 13-1. A broad T wave suggests LQT 1, a notched T wave LQT 2, and a long isoelectric ST segment and T wave, LQT 3. |
Our strategy for the diagnosis of the congenital LQTS is based on a careful personal and family history and close inspection of the resting ECG:
In patients with a history of presyncope, syncope, or SCA, the clinical details of the event are carefully examined to discern the likelihood of a ventricular arrhythmia.
A careful family history is obtained, asking specifically about SCD at an early age in first-degree relatives.
The ECG is examined for the duration of the QTc interval and the morphology of the T wave. ECGs from first-degree family members should also be obtained when possible.
If the clinical history suggests arrhythmic syncope and the QTc is significantly prolonged (i.e., >480 msec), no further testing is necessary to give the diagnosis of LQTS. Similarly, if the QTc is normal and the history is benign, no further testing is necessary to exclude the diagnosis.
In many cases, the diagnosis will remain uncertain, owing to an equivocal personal or family history and/or a borderline QTc. In such cases, an exercise test with assessment of microvolt TWA should be performed. If the QT interval fails to shorten appropriately during exercise or if TWA is present, the patient is considered to have LQTS (see Fig. 13-2).
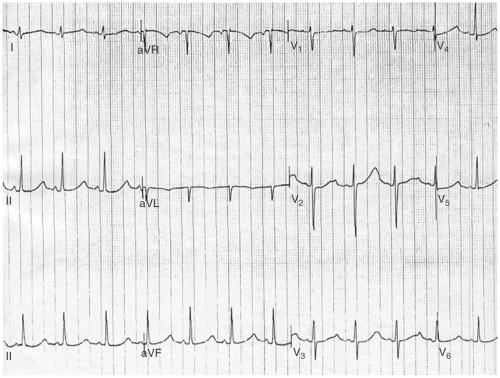 FIGURE 13-2. ECG demonstrating characteristic T wave alternans associated with the long QT syndrome (LQTS). |
Additional tests including electrophysiology study (EPS), pharmacologic provocation (e.g., epinephrine challenge), and facial immersion do not have established diagnostic accuracy and are not routinely included in the diagnostic evaluation for LQTS.
Genetic testing is currently commercially available to identify the most common genes involved in LQTS. When feasible, the patient of interest and first-degree family members with suspicious phenotypes (ECGs) should be genotyped.
Management of Congenital Long QT Syndrome
The common components in the management of patients with LQTS include:
Activity restriction: All patients with a diagnosis of LQTS should refrain from competitive athletics and from vigorous recreational sporting activities.
β-Blockers: The authors recommend β-blocker therapy in all patients with a diagnosis of LQTS regardless of whether or not symptoms are present. They have shown significant decrease in the rate of recurrent syncope and sudden death but are not effective enough in symptomatic patients to obviate the use of an implantable cardioverter defibrillator (ICD). β-Blockers should be titrated to blunt the maximal heart rate during exertion. It would be expected that these agents would be particularly useful in LQT 1 and less beneficial in LQT 3.
Potassium supplementation, particularly in LQT 2, has demonstrated a reduction in QTc duration but data pertaining to clinical events is not yet available. It is prudent to keep the potassium replete in patients with LQTS.
Mexiletine has been proposed for patients with LQT 3. Once again, clinical validation for this approach does not yet exist.
Sympathetic denervation: Left cardiac sympathetic denervation involves removal of the left stellate ganglion. This approach has been shown to reduce but not eliminate the risk of recurrent cardiac events and is associated with the development of Horner syndrome. It is rarely performed in the current era.
ICD implantation: Comprehensive indications for ICD implantation in patients with LQTS have not been defined. ICD therapy is appropriate for the secondary prevention of SCD in all LQTS patients with a history of SCA. In addition, the authors recommend ICD implantation in patients with recurrent syncope or TDP despite β-blocker therapy. Finally, ICD implantation is reasonable in selected high-risk patients, such as those with a strong family history of SCD or those with JLN.
Acute Management of Acquired Long QT Syndrome
Discontinuation of causative agent
Magnesium sulfate: 2 g bolus of 50% magnesium sulfate over 2 minutes
Consider isoproterenol to accelerate sinus rate and shorten the QT interval
Sodium bicarbonate if quinidine-related TDP
Consider placement of temporary pacing system with pacing at >80 bpm
Short QT Syndrome
The short QT syndrome is a recently described disorder of SCD in association with a QTc of <300 msec. The frequency of this disorder is unknown. At present, genetic abnormalities involving an increase in function of KVLQT1, HERG, and a decrease in function of Ik1 have all been described in association with a shortened action potential duration and surface QT interval. In addition to a short QT interval, there is relative absence of the ST segment and symmetric tall T waves in the precordial leads.
There is no role for EP testing in this disorder and the current management is an ICD in symptomatic individuals. The role for an ICD in first-degree relatives of symptomatic individuals who have the phenotype but no symptoms is uncertain.
Brugada Syndrome
The BS is a primary electrical disorder characterized by an ECG pattern of a pseudo-right bundle branch block with ST elevation in the right precordial leads (V1 to V3) and a history of documented or suspected ventricular arrhythmias and/or a family history of the disorder. Both clinical and ECG features are necessary to make a diagnosis of BS.
BS displays an autosomal dominant inheritance pattern. A single gene, encoding the cardiac sodium channel SCN5A, has been linked to BS. However, mutations of the SCN5A gene have been found in only 18% to 30% of BS families, and the genetic defect is unknown in most cases.
Most of the available epidemiologic data describe only the prevalence of Brugada ECG abnormalities. As the ECG findings are insufficient for the diagnosis of BS, and the penetrance of disease is incomplete, these data do not reflect the prevalence of either the clinical syndrome or the underlying genetic defect. The prevalence of a typical (type 1 see—subsequent text) Brugada-type ECG varies in different ethnic populations, ranging from up to 0.16% in Asian populations to <1 in 3,000 in a European cohort.
BS occurs in men more frequently than women.
The average age at diagnosis is late 30s or early 40s.
Events are not typically exercise induced and may occur during sleep.
The ECG manifestations may represent an early form of ARVD.
The pathophysiologic model of BS is based on reduced function of the cardiac sodium channel. Reduction of the inward sodium current during phase 0 of the myocardial action potential reduces the depolarization voltage peak. This defect in turn affects the impact of the repolarizing transient potassium
current I(to) in phase 1. Because the phase 0 peak is lower than normal, I(to) can drive phase 1 voltage lower below the level necessary to activate the calcium channels that maintain depolarization during phase 2. Without the influence of these calcium currents, the phase 2 “dome” of the action potential is lost, resulting in a dramatic shortening of action potential duration. Because I(to) is more prominent in the epicardium and M cells than it is in the endocardium (particularly, in the right ventricle), the impact of the sodium channel defect varies between myocardial layers. Therefore, a transmural voltage gradient develops during phase 2, producing the characteristic ST elevation in the right precordial leads. This heterogeneous loss of the phase 2 action potential “dome” can also produce the electrophysiologic substrate for arrhythmias through a process called phase 2 reentry. A full description of phase 2 reentry is detailed elsewhere, but is beyond the scope of this text.
current I(to) in phase 1. Because the phase 0 peak is lower than normal, I(to) can drive phase 1 voltage lower below the level necessary to activate the calcium channels that maintain depolarization during phase 2. Without the influence of these calcium currents, the phase 2 “dome” of the action potential is lost, resulting in a dramatic shortening of action potential duration. Because I(to) is more prominent in the epicardium and M cells than it is in the endocardium (particularly, in the right ventricle), the impact of the sodium channel defect varies between myocardial layers. Therefore, a transmural voltage gradient develops during phase 2, producing the characteristic ST elevation in the right precordial leads. This heterogeneous loss of the phase 2 action potential “dome” can also produce the electrophysiologic substrate for arrhythmias through a process called phase 2 reentry. A full description of phase 2 reentry is detailed elsewhere, but is beyond the scope of this text.
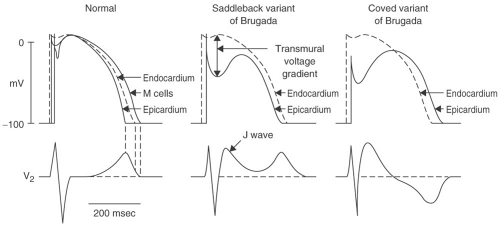 FIGURE 13-3. Proposed electrophysiology of the Brugada ECG demonstrating the mechanism of the saddleback and coved variants. (Adapted with permission from Scheinman MM and Keung E. J Am Coll Cardiol. 2007;49:2061–2069.) |
Brugada Diagnosis and Management
A Brugada-type ECG is not sufficient to make a diagnosis of BS; clinical features are required. Three types of ST-T wave abnormalities have been described in patients with BS. These findings must be present in at least two of the right precordial leads to qualify for the diagnosis of a Brugada-type ECG. The classic pattern, or type 1 ECG, has downsloping ST elevation of at least 2 mm with inverted T waves. This pattern is referred to as the coved-type Brugada ECG. In type 2 and type 3 Brugada ECGs, the elevated ST segment, after initially sloping downward, reflects upward into an upright T wave, referred to as a saddleback configuration (see Fig. 13-3). The distinction between type 2 and 3 ECGs is based on the magnitude of this ST elevation; ≥1 mm
for type 2 and <1 mm for type 3. Attention must be placed to the positioning of the precordial leads (particularly, V1 and V2) because placement of these leads in inappropriately high interspaces can cause a pseudo-Brugada pattern. In a consensus report from the Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology, it was proposed that the diagnosis of BS should be strongly considered in patients who meet the following criteria:
for type 2 and <1 mm for type 3. Attention must be placed to the positioning of the precordial leads (particularly, V1 and V2) because placement of these leads in inappropriately high interspaces can cause a pseudo-Brugada pattern. In a consensus report from the Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology, it was proposed that the diagnosis of BS should be strongly considered in patients who meet the following criteria:
A type 1 ST Brugada ECG plus at least one of the following clinical features:
Documented ventricular fibrillation (VF)
Self-terminating polymorphic ventricular tachycardia (PMVT)
Family history of SCD in those younger than 45 years
Type 1 ST segment elevation in family members
Electrophysiologic inducibility of VT
Unexplained syncope suggestive of a tachyarrhythmia
Nocturnal agonal respiration
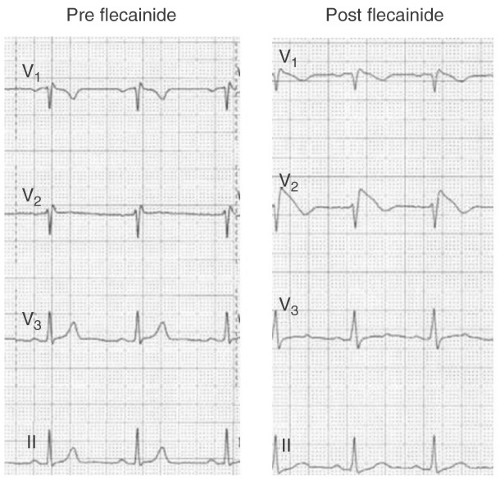 FIGURE 13-4. Conversion of Brugada type 2 ECG to a type 1 pattern following the administration of 300 mg of flecainide. |
Among patients with a type 2 or 3 ECG, drug challenge with a sodium channel blocker (e.g., flecainide 400 mg PO, or procainamide 10 mg per kg IV over 10 minutes) can convert the ECG to a type 1 pattern (see Fig. 13-4). According to many experts, and the ESC consensus statement, patients with
a type 2 or 3 ECG should only be diagnosed with BS if the ECG converts to type 1 following drug challenge. However, drug challenge is probably unnecessary in patients with a type 2 or 3 ECG who have documented VF, self-terminating polymorphic VT, unexplained syncope suggestive of a tachyarrhythmia, or nocturnal agonal respiration (clinical features a, b, f, and g). Most such patients will receive an ICD regardless of the results of drug testing.
a type 2 or 3 ECG should only be diagnosed with BS if the ECG converts to type 1 following drug challenge. However, drug challenge is probably unnecessary in patients with a type 2 or 3 ECG who have documented VF, self-terminating polymorphic VT, unexplained syncope suggestive of a tachyarrhythmia, or nocturnal agonal respiration (clinical features a, b, f, and g). Most such patients will receive an ICD regardless of the results of drug testing.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


