Sign/symptom
Differential diagnosis for SCA
Respiratory symptoms
Hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy, left ventricular outflow tract obstruction (LVOTO), restrictive or LV noncompaction cardiomyopathy
Chest pain
HCM, coronary anomalies, LVOTO, aortic rupture/Marfan, mitral valve prolapse (MVP), postoperative congenital heart disease
Seizures, syncope
Long QT syndrome, WPW, Brugada syndrome, CPVT, short QT syndrome, complete heart block
Palpitations
HCM, DCM, myocarditis, MVP, arrhythmogenic right ventricular cardiomyopathy, LQTS, WPW, Brugada syndrome
Drowning/near drowning
LQTS, CPVT
SIDS
LQTS, Brugada syndrome, CPVT, SQTS
Febrile seizures
Brugada syndrome
Congenital deafness
LQTS
The incidence of SCD in young persons is unknown and difficult to accurately estimate because of its rarity, marked differences among different age groups, and inconsistent reporting methods. Published retrospective and prospective studies estimate annual incidence rates of SCD in children and adolescents ranging from 0.8 to 6.2 per 100,000 patient-years. The risk is many times higher under 1 year of age. An estimated 4,000–8,000 children (≤18 years) in the USA die from SCD annually, primarily victims of sudden infant death syndrome (SIDS). SCD in young athletes is rare. Reported incidence rates in the USA range from 1 to 10 per 100,000 patient-years. The risk in male athletes is two- to fivefold higher than females. There also appears to be a higher incidence in African-American athletes in comparison to white athletes.
In most cases of SCD in children and young competitive athletes, a specific, often congenital associated underlying cardiovascular abnormality, cardiovascular can be determined (Tables 21.2 and 21.3). The causes vary with age. In older children and young athletes in the USA, hypertrophic cardiomyopathy (HCM) is the most frequent cause. In infants, coronary artery anomalies are the most common. In many infants, no structural cause can be identified, even after a thorough review of the history and death scene and a complete autopsy. This later group, by definition, is sudden infant death syndrome (SIDS). In patients less than 21 years of age, the most frequent causes are HCM, channelopathies, particularly the long QT syndromes (LQTSs), coronary artery abnormalities, and known, preexisting, structural congenital heart disease.
Table 21.2
Causes of SCD in the young
Condition | Types |
|---|---|
Myocardial abnormalities | Myocarditis |
Primary electrical disorders | Primary ventricular tachycardia/fibrillation |
Complete heart block, congenital and surgical | |
Commotio cordis | |
Coronary abnormalities | Anomalous origins from the aorta |
Anomalous origins from the pulmonary artery | |
Ostial abnormality | |
Myocardial bridge (tunneled) | |
Kawasaki disease | |
Transplant vasculopathy | |
Cocaine vasospasm | |
Coronary atherosclerosis | |
Congenital heart disease | Aortic stenosis |
Coarctation of the aorta | |
Tetralogy of Fallot (including variants) | |
Transposition of the great arteries | |
Atrioventricular discordance | |
Hypoplastic left heart syndrome | |
Single ventricle/Fontan | |
Ebstein’s anomaly | |
Truncus arteriosus | |
Miscellaneous | Primary pulmonary hypertension |
Marfan’s syndrome | |
Eisenmenger syndrome | |
Mitral valve prolapse |
Table 21.3
Genetic causes of sudden cardiac death
Hypertrophic cardiomyopathy |
Arrhythmogenic right ventricular cardiomyopathy |
Marfan syndrome (aortic rupture) |
Dilated cardiomyopathy/restrictive cardiomyopathy |
Long QT syndrome |
Brugada syndrome |
Catecholamingergic polymorphic ventricular tachycardia |
Short QT syndrome |
Primary pulmonary hypertension |
Sudden Infant Death Syndrome
SIDS is a diagnosis of exclusion, loosely defined as unexpected death under a year of age that occurs during sleep and remains unexplained after thorough clinical history and postmortem examination (standard autopsy). The incidence is relatively high but has decreased to less than 1/1,000, at least in part related to well-publicized supine (on the infant’s back) sleeping recommendations. SIDS remains a leading cause of infant death in developed countries. It is generally accepted that SIDS deaths occur as a result of a variety of causes, including metabolic, environmental, and immunologic.
A number of genetically mediated arrhythmias have been postulated as a probable cause of SIDS. To date, mutations in 16 cardiac ion channel genes have been identified in victims of SIDS, most of them known to be functionally significant or disease causing. The spectrum of channelopathy genes represented in SIDS cases includes those associated with many forms of the LQTS, the Brugada syndrome, the short QT syndrome, and catecholaminergic polymorphic ventricular tachycardia (CPVT). A minority of the cardiac ion channel gene defects associated with SIDS have not been established to be functionally significant or malignant. On the other hand, there is evidence that some isoforms, as a result of alternative splicing of affected may behave more malignantly in fetuses and young infants. To what extent LQTS contributes to SCD is unclear. Early series implicated LQTS as a cause of SCD in a relatively low percentage, but SCD in the young is often unexplained after a standard autopsy. With postmortem genetic testing (molecular autopsy) becoming more commonplace, it appears that LQTS is the cause of SCD in the young in 10–20 %, potentially more in younger patients (see Chap. 19).
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM; see Chap. 19) is a diverse disease in its genetic, biochemical, cellular, phenotypic, and clinical features. In infants and children, HCM phenotype can be caused by inborn errors of metabolism, neuromuscular disorders, and malformation syndromes, but in the majority of cases it is a result of a mutation in 1 of at least 20 genes that encode protein elements of the cardiac sarcomere. These mutations are inherited, as autosomal dominant with variable penetrance, similar to most defects in structural proteins. With an estimated prevalence of 1 in 500, it is the most common inherited cardiovascular disease. Despite early observations suggesting a high risk in all patients with HCM, it does not uniformly herald an unfavorable prognosis; overall, the annual mortality is about 1 %, with a near normal life expectancy and little morbidity in most patients. There exists, however, a subset of patients, 10–20 % of those with HCM, in which the annual mortality from SCD exceeds 5 %.
A sudden arrhythmia is the most frequent means of sudden death, often as the first clinical manifestation of the disease, and typically in association with exertion. It occurs most commonly in young adults, adolescents, and children, a population with an estimated annual SCD of 2–5 %. Primary ventricular tachycardia and its degeneration to ventricular fibrillation is the predominant mechanism. Patchy and confluent myocardial scars, as a result of abnormal coronary architecture, diminished coronary flow reserve, coronary-myocardial mismatch and consequent myocardial ischemia, in addition to collagen infiltration, cellular disarray, dispersion of impulse conduction and refractory periods, and abnormal calcium handling serve as electrophysiologic substrates for the ventricular arrhythmias. Acute ischemia and vascular instability with hypotension are proposed triggers.
Major risk factors for SCD in adults with HCM are listed in Table 19.5. Most patients are free of risk factors and SCD in such patients is uncommon, although this has not been established in young patients. It is clear that adults with two or more major risk factors are at a substantially increased risk. Patients with no risk factors have been shown to have a 6-year SCD-free survival rate of 95 %, while the corresponding rate for those with two or more risk factors was 72 %.
Implantable cardioverter-defibrillator therapy is recommended for secondary prevention following events of resuscitated SCD and should be considered for primary prevention in those with at least one major risk factor. Patients and families should be well informed because ICD complication rates, including a high number of inappropriate shocks, are common in this population. There is consensus that individuals with HCM should be excluded from most competitive sports, with exception of those designated low static, low dynamic. Recommendations for genotype-positive–phenotype-negative individuals identified by cascade genetic screening are less clear. Studies are underway to help define the risk of exercise in this genotype-positive group but without ventricular hypertrophy on the echocardiogram.
Arrhythmogenic Right Ventricular Cardiomyopathy (See Chap. 19)
Arrhythmogenic right ventricular cardiomyopathy (ARVC), formerly arrhythmogenic right ventricular dysplasia, is characterized by fibro-fatty replacement of the right ventricular myocardium with progressive right ventricular dilation and dysfunction and less evident involvement of the left ventricular myocardium. The prevalence is estimated to be 1/2,000 to 1/5,000, but an associated genetic abnormality may be as prevalent as 1/200. It appears that at least 50 % have genetic abnormalities of desmosome proteins. Intense endurance sports may lead to a similar phenotype or, more likely, vigorous exercise may increase penetrance and risk of arrhythmia. ARVC may result from an inflammatory or remodeling process adversely modulated by genetic alterations in desmosome proteins.
ARVC most often presents with SCA in adolescents and young adults and is more common in males. Inheritance is autosomal dominant with variable penetrance and about 30 % have a positive family history. In Italian series of SCD, roughly ¼ have ARVC. This is in contrast to less than 2 % in series from the USA. The diagnosis can be challenging to make and standard echocardiography is not sensitive in the absence of a high index of suspicion. Cardiac magnetic resonance and electro-anatomic and voltage mapping have been shown to be more sensitive. ECGs are abnormal in about 80 %, characterized by T wave inversion and epsilon waves and/or delayed S upstroke in V1 − V3/V4, and low voltages in limb leads. Age-related changes in the T wave polarity in the right precordial leads (so-called juvenile T waves) can confound the diagnosis; the usual negative T waves seen in preadolescence revert to a positive direction at ages 12–18 years (Fig. 21.1). Italian series have reported success in reducing the number of SCD in young athletes using screening programs, largely attributed to identification and subsequent strict activity limitation of individuals with ARVC.
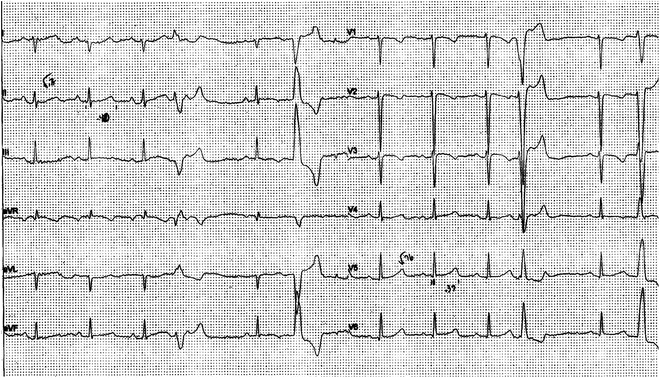
Fig. 21.1
Premature ventricular beats (multiform) and negative T waves in V2–V3 in an asymptomatic 17-year-old girl suggesting ARVC. She was genotype positive for a plakophilin mutation
Myocarditis
Myocarditis is an inflammatory process of the myocardium. There are a multitude of etiologies, including drugs, toxins, autoimmune diseases, Kawasaki disease, rheumatic fever, and a wide variety of infectious pathogens. The majority of cases are caused by viral infections, most commonly Coxsackie group B enteroviruses, adenoviruses, human herpesvirus 6, and parvovirus B19. Viral cytopathic injury followed by immune-mediated myocardial damage results in myocyte necrosis and diffuse inflammatory infiltrates. This and subsequent patchy fibrosis and persistence of a low-grade inflammatory state can serve as arrhythmogenic substrates for ventricular tachycardia (Fig. 21.2). Myocarditis can be subclinical and is a well-recognized cause of acute heart failure and subsequent dilated cardiomyopathy, but it is probably under-appreciated as a cause of SCD. A recent study of all autopsy referrals to a single center in the UK showed 5 % of all pediatric deaths over age 5 were secondary to myocarditis and more than half of those were sudden. Ventricular ectopy or SCD may be the initial clinical manifestation of acute myocarditis and can occur in the presence of preserved ventricular function, in some cases confounding the diagnosis. Treatment is largely supportive. Recovery is complete in most, but a minority may experience SCD, develop chronic dilated cardiomyopathy or are lost due to fulminant heart failure.
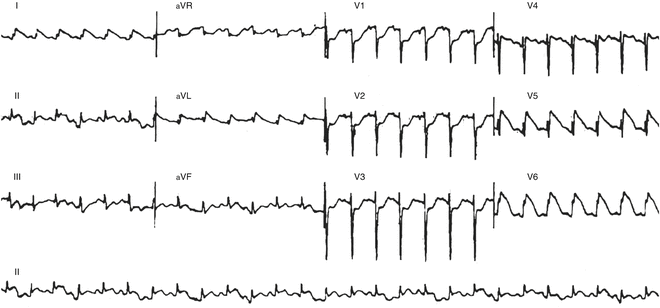
Fig. 21.2
Sinus tachycardia with ST elevation in a 10-month-old with myocarditis (autopsy proven)
Congenital LQTS (See Chap. 19)
Congenital long QT syndrome (LQTS) (Chap. 19, Figs. 19.1–19.4) represents a spectrum of channelopathies that share in common the prolongation of ventricular repolarization and clinical manifestations of syncope and SCA, often associated with physical exertion or high adrenergic states. The estimated prevalence is 1:2,000–2,500. Inheritance is autosomal dominant in the majority of cases (Romano–Ward syndrome). The more severe Jervell and Lange-Nielsen syndrome is associated with congenital deafness.
The prolongation of repolarization can lead to delayed after-depolarization-mediated polymorphic ventricular tachycardia, torsades de pointes (TdP). TdP (Fig. 21.3), translated as twisting of points, is a distinctive, uncommon form of rapid VT characterized by a variable QRS amplitude and axis that appear to twist around a baseline. TdP is often self-limiting producing syncope, seizures, dizziness, or fleeting palpitations; but it can degenerate into ventricular fibrillation and result in SCA or SCD.
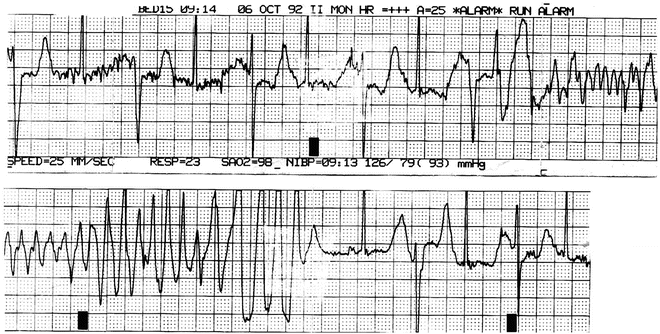
Fig. 21.3
ECG strip forma 16-year-old girl with TdP. She had a long QT on her resting ECG. Genetic testing was not available
Arrhythmia triggers, to an extent, have been shown to be gene specific. Individuals with LQT1 are at risk during exercise or emotional stress. Swimming-related events in particular have been closely linked to LQT1. Events triggered by sudden or loud noises have been linked to LQT2. Individuals with LQT3 are at higher risk while asleep or at rest. Risk stratification is challenging largely because of the significant degree of inter-gene and intra-gene variability, in addition to incomplete penetrance and variable expressivity. Risk is clearly and most reproducibly increased in the presence of very long corrected QT intervals, irrespective of gene defect. A QTc of ≥500 ms is an indicator of high risk. The risk is higher in children, infants, and fetuses. The risk appears to be higher in female patients with LQT2 and slowly but significantly increases with age while decreasing in males. Male patients with LQT3 are at higher risk. The risk may be lower in individuals with LQT1, possibly because a larger percentage of KCNQ1 genotype-positive individuals have normal QT intervals (incomplete penetrance).
The genetic and phenotypic heterogeneity contributes to challenges in diagnosing and managing patients with LQTS. Importantly, it makes excluding the diagnosis challenging as well. There are wide ranges of corrected QT interval values in unaffected and affected individuals, even among individuals with the same gene defect. Age-related variations in conduction system properties, heart rates, and sinus arrhythmia also confound the diagnosis, particularly in the absence of symptoms or family history. Interpreting QT intervals should be done in context. A QTc of 450 ms in an individual with a genetically confirmed first-degree relative or recurrent exertional syncope has a higher predictive value than a QTc of 475 ms in an asymptomatic individual with no family history. In the absence of concerning symptoms or family history, QTc intervals of greater than 460–470 ms for males and 470–480 ms for females are reasonable thresholds to seriously consider the diagnosis. T-wave morphology can be helpful. Notched, biphasic, and very late peaking T-waves are suggestive; T-wave alternans is highly suggestive. Using the LQTS Clinical Probability Score (Chap. 19, Table 19.2) is helpful. Exercise stress testing, Holter monitoring, and pharmacologic challenge can be useful. Commercially available genetic testing can confirm the genetic diagnosis, screen family members, and to help guide clinical management (see Chap. 19).
Therapy includes a beta-blocker in almost all patients. Propranolol and nadolol have been shown to be more effective than metoprolol. Sodium channel blockers, such as mexiletine, are a useful adjunct in some patients with LQT3, but it is recommended that the QT shortening effect be tested because the response appears to be mutation dependent. Left cardiac sympathetic denervation has been shown to be effective in some cases, including those with breakthrough symptoms, those with very long QTc intervals, and those with Jervell and Lange-Nielsen syndrome or homozygous or compound heterozygous mutations. ICD implantation may be indicated for secondary prevention and other high risk situations. It is important that patients and families are counseled about avoidance of medications that cause QT interval prolongation and avoidance of potentially dangerous situations, such as unobserved swimming in those with LQT1 and alarms and startling noises in those with LQT2.
The 36th Bethesda Conference Guidelines (2005) and the European Society of Cardiology (2006) recommend against involvement in nearly all athletics for individuals with symptomatic or genetically confirmed LQTS in the presence of significant QTc prolongation. In asymptomatic individuals with borderline QTc prolongation and in genotype-positive–phenotype-negative individuals, appropriate guidance is not clear. As described, risk stratification is, at best, challenging and there is some evidence of very low rates of events in treated patients who choose to participate in athletics. There is also evidence that arrhythmias are less common in individuals with QTc intervals less than 500 ms. It is very likely that recommendations will continue to evolve.
Brugada Syndrome (See Chap. 19)
Brugada syndrome is characterized by SCD and the ECG findings of coved-type ST elevation in the right precordial leads and right bundle branch block (Chap. 19, Figs. 19.5 and 19.6). This type I Brugada ECG pattern is intermittent and may be unmasked by pharmacologic challenge with sodium channel blockers such as procainamide, ajmaline, or flecainide (the latter two are not available as IV preparations in the USA). This pattern is not specific and can be seen with electrolyte abnormalities or myocardial infarction. The diagnosis of Brugada syndrome requires, in addition to the Brugada ECG pattern, documented VT/VF, a history of agonal nocturnal breathing or unusual syncope, suggestive family history of SCD, or Brugada syndrome (or Brugada ECG pattern) in family members. The prevalence is unknown and estimates vary widely from 1/1,000 to 1/100,000; the diagnosis is often made based on ECG pattern only. There is a higher prevalence in certain populations or areas, notably in Southeast Asia.
Brugada syndrome is genetically heterogeneous and complex. Approximately 75 % of those phenotypically affected are male with manifestations typically occurring in the third, fourth, and fifth decade. Most arrhythmias occur at rest, but fever and consumption of large meals are established triggers.
Early reports suggested that Brugada syndrome was highly malignant, but more recent studies have concluded that the yearly incidence of arrhythmic events is between 0.4 and 4 %. Risk factors include persistent type I Brugada ECG pattern in the absence of provocation and history of syncope or SCA. There is debate as to the utility of invasive electrophysiology study to risk stratify asymptomatic patients. Implantable cardioverter-defibrillators are indicated for symptomatic patients or for secondary prevention. Quinidine has been shown to be effective in preventing arrhythmia recurrences and is being studied in asymptomatic individuals with Brugada. Interestingly, patients with Brugada syndrome with ICDs have a high rate of inappropriate shocks related to a high incidence of supraventricular tachyarrhythmias, most commonly atrial fibrillation. Given the relatively low rate of arrhythmic events and the high rate of inappropriate shocks, ICDs are no longer routinely recommended for primary prevention in individuals with Brugada syndrome.
Catecholaminergic Polymorphic Ventricular Tachycardia (Chap. 19)
Catecholaminergic polymorphic ventricular tachycardia is a rare heritable arrhythmia that is characterized by adrenergic-induced ventricular tachyarrhythmia and sudden death (see Chap. 19, Fig. 19.8). Patients with CPVT often develop symptoms of syncope and sudden death early in childhood. A great degree of variability is seen even among members of the same family who carry the same mutation. Symptoms usually occur with stress or exercise. Sudden death in early adulthood in symptomatic patients is a risk if the condition is left untreated.
Other Repolarization Abnormalities
Other repolarization abnormalities have been reported: the short QT syndrome (Chap. 19) comprises QTc < 320 ms, very tall T waves, and SCD. Management usually requires an ICD. Finally the emergence of other repolarization syndromes is less well defined but likely contributes to SCD in the young (Chap. 19).
Wolff–Parkinson–White Syndrome (See Chap. 4)
The prevalence of ventricular preexcitation or Wolff–Parkinson–White (WPW) ECG pattern is estimated to be 1–3/1,000. The risk of SCD in those with WPW is very low. Symptomatic patients have an estimated risk of approximately 0.25 % per year or 3–4 % over a lifetime. The risk in asymptomatic patients is lower, approximately 1 per 1,000 patient-years. The mechanism of SCD is rapid anterograde conduction across a fast-conducting accessory pathway(s) with short anterograde effective refractory period (ERP) during atrial fibrillation (Fig. 21.4).
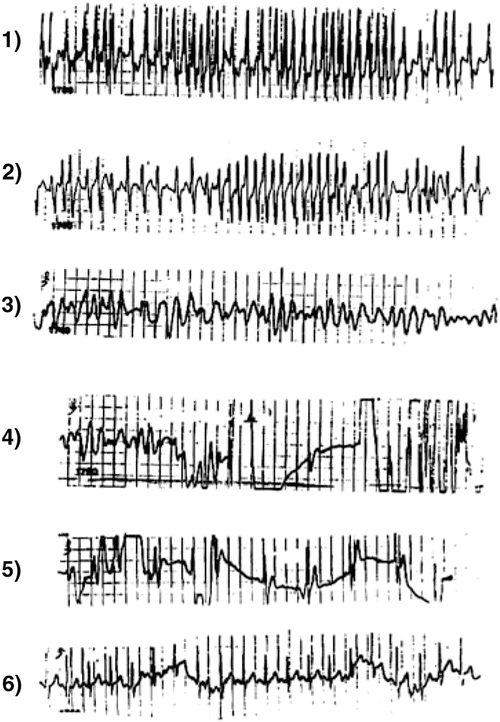
Fig. 21.4
Panels 1 and 2: Atrial fibrillation in a 9-year-old boy with a very rapid ventricular response deteriorating into ventricular fibrillation (Panels 3 and 4) and cardiac arrest. He was successfully defibrillated (Panels 4 and 5) into sinus tachycardia with no preexcitation (Panel 6). At electrophysiologic study had an intermittent anterograde left lateral accessory pathway with an effective refractory period of 190 ms. It was successfully interrupted by radiofrequency ablation
Risk factors for SCD include a short preexcited RR interval during atrial fibrillation or rapid atrial pacing (Fig. 21.4), a short anterograde ERP (<220–250 ms) of the accessory pathway, multiple pathways, inducible AVRT or history of AVRT, male gender, and history of syncope. Arrhythmia inducibility and young age are associated with a significantly increased risk of arrhythmic events, including SCD, in previously asymptomatic patients. However, because of a small atrial mass, atrial fibrillation is very uncommon in infants and young children.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


