Fig. 24.1
Percent change in age-adjusted death rates for coronary heart disease by race and sex, U.S., 1950–2008 (Source: National Heart Lung, and Blood Institute [9])
Population Dynamics and Sudden Cardiac Death
The epidemiologic data from the Framingham Heart Study, a 26-year follow-up of 5,209 men and women 30–59 years old and free of identified heart disease at baseline, showed that SCD accounted for 46 % of deaths due to CAD among men and for 34 % among women [12]. The incidence of SCD increased with age. However, the proportion of sudden, unexpected deaths from CAD was greater in the younger age groups. The 300,000 SCDs that occur annually in the United States can be expressed as a fraction among an unselected adult population. The overall incidence, therefore, is 0.1–0.2 % per year. When the high-risk subgroups are identified and removed from this population base, the calculated incidence for the remainder of the population decreases, and identifying individuals at risk becomes more difficult (Figs. 24.2 and 24.3). Based on these estimates, any preventive measure must be applied to 999 of the 1,000 individuals who would not have an event during the course of a year to potentially influence the outcome in 1 of 1,000. The costs of such a low-yield intervention are obviously prohibitive; therefore, we must identify more specific markers of high risk. The present risk factors generally identify the risk of developing structural heart disease rather than the proximate precipitator of the SCD event.
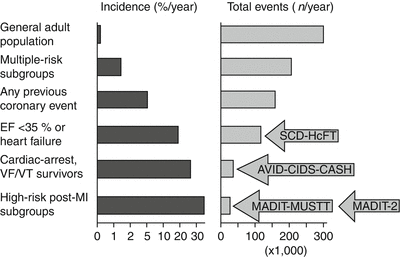
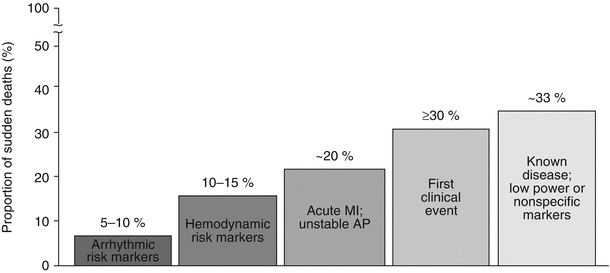
Fig. 24.2
Relationship between population subsets, incidence of sudden cardiac death, and total population burden for each group. With increasing incidence, based on subgroup profiling, there is a decreasing proportion of the total sudden death burden. This relates to the population impact of the outcomes of implantable cardioverter-defibrillator trials
Fig. 24.3
Subgroups of ischemic heart disease patients at risk for sudden cardiac death. The population subset with high-risk arrhythmia markers carries <10 % of the total sudden death burden attributable to coronary artery disease. A somewhat larger group is associated with hemodynamic risk markers and congestive heart failure. More than 50 % of the total sudden death burden is accounted for by those victims among whom sudden cardiac death is the first clinical event or those who have known coronary heart disease but low power of risk. AP action potential, MI myocardial infarction (From Myerburg [10]. © Futura Publishing Company, Inc. 2001. Reprinted with permission from John Wiley and Sons)
Time Dependence of Risk Factors
The risk of death after a major change in cardiovascular status is not linear over time in most clinical circumstances [13, 14]. The highest secondary death rate occurred during the first 6–18 months after a major event, such as cardiac arrest, new onset of heart failure, unstable angina, and recent myocardial infarction (MI). The slope of the survival curve approaches that of a similar population that has remained free of an interposed major event at 18–24 months [1, 13]. A recent study showed that the risk of SCD was highest (1.4 % per month) in the first month after MI and decreased to 0.14 % per month after 2 years [14].
Current practice guidelines recommend assessing left ventricular (LV) function 40 days after MI to determine the necessity of implantable cardioverter-defibrillator (ICD) implantation for primary prevention of SCD [8].
Risk Factors
In a study by Gillum [15] of persons between the ages of 35 and 74 years in 40 states, the epidemiology of SCD parallels that of CAD. The annual incidence of SCD was 1.91 per 1,000 for white and nonwhite men, 0.57 per 1,000 for white women, and 0.90 per 1,000 for nonwhite women (Fig. 24.4). In patients with ischemic heart disease, 60 % of deaths in men and 50 % in women occurred out of hospital. Another study [16] showed that the mean age of SCD victims was 70 years in men and 82.4 years in women. The cause of death for SCD differed for people 35–64 years old and people 65 and older. Acute ischemic heart disease, unspecified cardiovascular disease, cardiomyopathy, and dysrhythmias were more common in the younger group than in the older group. In contrast, the cause of death for SCD in the older group was more frequently chronic ischemic heart disease or heart failure.
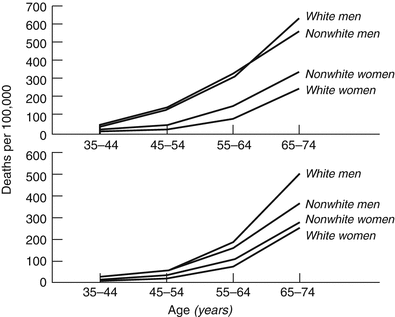
Fig. 24.4
Mortality rates for ischemic heart disease occurring out of hospital or in emergency departments (top) and occurring in hospital (bottom) by age, sex, and race in 40 states during 1985 (From Gillum [15]. Reprinted with permission from Wolters Kluwer Health)
According to the Heart Disease and Stroke Statistics 2013 Update, the rate of death due to cardiovascular disease declined by 32.7 % and was 236.1 per 100,000 in 2009. In addition, CAD alone accounted for 1 of every 6 deaths in the United States in the same period [6].
Influence of Age, Race, and Sex
Age
The incidence of SCD increases with age in men and women, both white and nonwhite, because the prevalence of ischemic heart disease increases with age. For people under 35, the peak incidences of SCD are between ages 25 and 35 years, if sudden infant death syndrome cases are excluded. For people 35 and older, SCD is most likely to occur between 75 and 85 years. The incidence of SCD is 2.28/100,000 person-years in persons <35, 4.40/100,000 person-years in the 25–35 group, and 100/100,000 person-years in adults older than 35 [5, 17]. However, the proportion of SCD caused by CAD decreased with age from approximately 75 % at ages 35–42 to approximately 50 % at ages 75–84 [16].
Race
Sudden cardiac death is more common among black Americans than white and Hispanic Americans. Hispanic Americans have lower SCD rates than non-Hispanic populations [18]. Moreover, survival rates after in-hospital cardiac arrest are lower for black American patients [19]. A large study in Chicago concluded that the incidence of cardiac arrest was significantly higher for African Americans than for white Americans in every age group [20]. The survival rate after cardiac arrest was 2.6 % in white patients, compared with 0.8 % in black patients. When African Americans had cardiac arrest, the event was significantly less likely to be witnessed than when white or Hispanic patients had cardiac arrest. Likewise, black patients who had SCD were significantly less likely to receive standard-initiated cardiopulmonary resuscitation or have a favorably initial rhythm on admittance to the hospital than white or Hispanic patients with SCD. When black patients were admitted, they were half as likely to survive and had lower rates of postresuscitation care than white patients [19, 20].
Sex
The annual incidence of SCD is three to four times higher in men than in women; approximately 75 % of SCDs occur in men. This can be explained by the higher incidence of CAD in men than in women and the protection from atherosclerosis that estrogen gives to premenopausal women. After 20 years of follow-up in the Framingham Heart Study, there was a 3.8-fold excess incidence of SCD in men compared with women. The excess rate in men peaked at 6.75:1 in the 55–64 age group and then fell to 2.17:1 in the 65–74 age group [11].
Physical Activity
Heavy exercise can trigger the onset of an acute MI, particularly in persons who are habitually sedentary. A prospective case crossover study [21] showed that the relative risk of sudden death associated with an episode of vigorous exertion was lower among those who exercised more frequently. Men who rarely engaged in vigorous exercise (less than once a week) had a relative risk of sudden death: 74.1 during and 30 min after exertion. In comparison, men who exercised at least five times per week had a relative risk of 10.9, which was much lower. However, this risk was still significantly higher than during periods of lighter exertion. Despite the high relative risk, the absolute excess risk of sudden death during any particular period of vigorous exertion was still extremely low and similar to that reported in other populations. It has been suggested that vigorous exercise increases platelet adhesiveness and aggregability, whereas moderate physical activity may be beneficial by decreasing platelet adhesiveness and aggregability [22]. Furthermore, acute exercise stresses the sympathetic nervous system and decreases vagal activity, which can lead to an acute increase in susceptibility to ventricular fibrillation [23]. In contrast, regular vigorous exertion increases vasovagal tone, resulting in increased cardiac electrical stability and protection against ventricular fibrillation [24]. Cardiac arrest occurs at a rate of 1 per 12,000–15,000 during rehabilitation programs, whereas during stress testing, cardiac arrest occurs at a rate of 1 per 2,000. This is at least six times greater than the incidence of SCD for patients known to have heart diseases [2]. Other data indicate that the effect of activity on SCD may be small. The national sports-related sudden death study in France showed that the incidence of exertion-related SCD was between 5 and 17 cases per million per year [25].
Psychosocial Factors
Social isolation, stress at work, depression, anxiety, marital problems, death of a child, caring for a sick spouse, and childhood abuse have been associated with an increased risk of cardiovascular mortality [26]. Rahe and associates [27] reported a correlation between an increased life change score in the preceding 6 months and the risk of coronary events. This association was particularly notable for victims of SCD. A study of SCDs among women showed an increased risk for women who were not married, who had fewer children than the control group, and who had a greater educational discrepancy with their spouses than did age-related controlled subjects living in the same environment [28]. Other risk factors in this group included prior psychiatric treatment, greater alcohol consumption than the control group, and cigarette smoking [28]. In a large study of 2,320 men who survived MI, social isolation and high stress were associated with an increased risk of SCD. Both of these factors were directly associated with low educational levels [29]. Type D personality has also been associated with an increased incidence of ventricular arrhythmia and SCD [30]. A study of SCD at the time of an earthquake reported SCD triggered by an earthquake [31].
Risk Factors for Coronary Artery Disease, Ischemic Heart Disease, and Sudden Cardiac Death
The risk factors for SCD parallel those of CAD, the most common cause of SCD in developing countries. Coronary artery disease accounts for 80 % of all SCD, and risk factors such as family history of CAD, hypertension, diabetes mellitus, male sex, old age, cigarette smoking, hypercholesteromia, kidney dysfunction, and obesity increase incidence of CAD and SCD [4, 11, 32]. Sudden cardiac death is the first clinical manifestation of CAD in more than 30 % of patients with CAD (Fig. 24.3) [14].
A prior history of CAD is a powerful risk factor for SCD. In a review of SCD in the Framingham study, the risk of SCD was 3–12 times higher among those with clinical manifestation of CAD than among the general population of the same age. In men, the risk was on average 6.7 times that of persons without a CAD event. The risk of SCD was higher in persons with an MI than in those who had angina pectoris. However, even angina carried an almost fivefold increased risk [33]. Of the risk factors discussed above, diabetes, kidney disease, and smoking are specific risk factors for SCD after the onset of coronary artery disease, and the rest correlate only with CAD [34]. In persons with established CAD, factors that reflect ischemic myocardial damage were the chief predictors of sudden death. Electrocardiographic abnormalities indicating old MI, left ventricular hypertrophy (LVH), intraventricular conduction, or repolarization abnormality were significant predictors of SCD (Fig. 24.5).
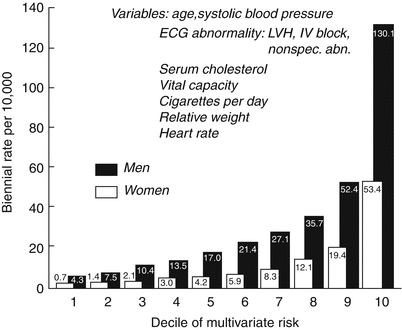
Fig. 24.5
Risk of sudden cardiac death by decile of multivariate risk: 28-year follow-up, Framingham Study. ECG electrocardiographic, IV interventricular, LVH left ventricular hypertrophy, Nonspec. abn. nonspecific abnormality (From Kannel and Schatzkin [341]. Reprinted with permission from Elsevier Limited)
The principal mechanism by which hypertension predisposes to SCD is via LVH. Other factors that influence the development of LVH include age, obesity, stature, glucose intolerance, and genetic factors [35–38]. The greater prevalence of hypertension in black men than in white men may explain the greater incidence of SCD despite the lower prevalence of CAD [39]. The presence of electrocardiographic LVH as manifested by increased voltages and repolarization abnormalities was associated with a 5-year mortality of 33 % in men and 21 % in women [35, 38]. The risk of SCD in the presence of electrocardiogram (ECG) findings of LVH was comparable to that of CAD or heart failure. Left ventricular hypertrophy has become less prevalent during the past four decades, which has coincided with improved hypertension control. However, people treated for hypertension still have a higher risk of SCD than those not treated for hypertension, even after correction for achieved blood pressure [36]. Left ventricular hypertrophy identified by an echocardiogram or ECG contributes independently to cardiovascular risk, and the presence of LVH identified by both confers a greater risk than LVH identified by either alone [35]. Other electrocardiographic findings could also be helpful in identifying patients with an increased risk of SCD, including the presence of intraventricular conduction delays, QT prolongation, and an increase in the resting HR of survivors of out-of-hospital cardiac arrest [2].
However, studies by Zabel and colleagues [40] failed to support the usefulness of QT dispersion in survivors of out-of-hospital cardiac arrest. In survivors of cardiac arrest of unknown cause who have a left ventricular ejection fraction (LVEF) <30 %, the risk of SCD exceeds 30 % if they do not have inducible ventricular tachycardia (VT) by programmed extra stimulation for 1–3 years [2, 41–43]. In those with inducible VT, the risk of recurrent arrest ranges from 15 to 50 % over 2–3 years, despite amiodarone therapy or therapy that suppresses inducible arrhythmia [41, 43–45].
Left ventricular ejection fraction is a strong predictor of subsequent SCD. Despite less than optimal sensitivity and specificity, LVEF is the cornerstone of decision-making regarding primary prevention of SCD [46]. Patients with LVEF of less than 30–35 % are considered high risk and qualify for primary prevention with ICD [47].
Different multivariable risk algorithms to identify individuals with high SCD risk have been created based on analyses of large clinical trials. The main factors in these algorithms are age, functional class, LVEF, LV conduction abnormalities, nonsustained VT, and history of heart failure. These algorithms need to be validated by large prospective studies [4, 48].
In fact, lack of specificity is a limitation among all individual SCD risk markers, including LVEF [4], and developing a more accurate technique to assess risk of SCD is crucial. New risk stratification techniques, such as myocardial substrate imaging by cardiac magnetic resonance imaging (MRI) and magnetic field imaging for prediction of arrhythmic events, have had promising results, but further prospective data are necessary to validate these approaches [46, 49].
Transient Risk Factors
Transient risk indicates a time-limited and unpredictable event or state that could initiate or enable the initiation of an unstable electrophysiologic (EP) condition. It increases the probability of transition from normal to benign cardiac rhythm to VT or VF [1]. Unfortunately, these risk factors are transient and lack sufficient sensitivity, specificity, and predictive values to be used for a specific preventive or therapeutic intervention before an actual event.
Transient Ischemia and Reperfusion Arrhythmias
Ischemia has a clear clinical correlation with potentially fatal ventricular arrhythmias during the early phase of an acute MI. However, approximately 80 % of SCDs caused by CAD are not associated with an acute MI. It is assumed that transient acute ischemia is one of the major triggering factors of SCD [50]. A study in an experimental model showed that smaller decreases in blood flow are required to induce VT or VF in the presence of MI than in controlled subjects without prior infarct [51]. Reperfusion can also induce electrical instability [52] via reentry or triggered activity mechanisms [53].
Systemic Factors
Reversible systemic abnormalities could contribute to life-threatening arrhythmias. Imbalances in electrolytes such as hypokalemia, hypomagnesemia, hypoxemia, and acidosis may influence EP stability and cause VT/VF and SCD. Recognizing and correcting these factors are the only required interventions. Hemodynamic dysfunction in patients with an abnormal heart can result in cardiac arrest. In an experimental model, volume loading of isolated perfused canine left ventricles shortened the refractory periods [1, 54], and regional disparity in hearts with prior MI has been shown [1, 54].
Autonomic Variation
Altered HR due to autonomic imbalance raises the risk of SCD among survivors of MI [55] and survivors of out-of-hospital cardiac arrest [56]. A blunted baroreceptor response to phenylephrine has also been suggested as a marker for the risk of VT or SCD after MI [1, 57]. That isoproterenol can induce sustained VT and beta blockers can prevent sustained VT in survivors of sudden cardiac arrest suggests that autonomic influence has a role in creating ventricular arrhythmias. Uikuri and associates [58] studied the sinus node rate as an estimate of cardiac autonomic tone immediately after the onset of VT. Sinus node rate during ventricular atrial dissociation increased progressively during the first 30 s of VT in patients with stable VT. However, in patients with unstable VT, the sinus node rate increased more rapidly during the first 5 s and then abruptly decreased [1, 58]. All of these observations indicate that an abnormal autonomic function is a risk factor for VT/VF and SCD. These markers provide information about autonomic balance. The risk is usually increased when there are signs of reduced vagal activity to the heart. The concept that an elevated heart rate (HR) increases the mortality risk was validated by the Gruppo Italiano per lo studio della Sopravvienza nell’ Infrato Miocardico (GISSI-2) study [59]. In 8,915 post-MI patients, HR at hospital discharge was an independent predictor of total mortality. In this study, SCD represented almost 50 % of all mortality.
The Autonomic Tone and Reflexes After Myocardial Infarction (ATRAMI) study [60], which enrolled 1,284 patients with a recent (less than 28 days) MI, provided prospective data on the additional and independent prognostic value for cardiac mortality of heart rate variability (HRV) and baroreflex sensitivity. The ATRAMI investigators showed that after MI, the analysis of autonomic markers has significant prognostic value independent of established clinical predictors, such as EF and ventricular arrhythmias. These investigators showed that, during 21 months of follow-up, depressed heart rate variability and baroreceptor sensitivity were significantly associated with cardiac mortality (odds ratio, 3.2 and 2.8, respectively). The combination of low HRV and depressed baroreflex sensitivity further increased the risk. In this study, 1-year mortality increased from 1 % when both markers were well preserved to 15 % when both were depressed. Furthermore, the association of LVEF <35 % with low HRV further increased the risk; risk rose even more when low baroreflex sensitivity was also associated with LVEF <35 % and low HRV. However, in patients older than 65, the predictive power of baroreflex sensitivity declined much more markedly than heart rate variability. For this reason, this specific prognostic value was higher for baroreflex sensitivity in patients younger than 65 and for heart rate variability in patients older than 65.
A recent study showed that there is a strong relationship between increased resting HR and SCD in the general population, even after adjustment for left ventricular systolic dysfunction and HR-modulating drugs. This finding implies that therapies that modulate autonomic tone could prevent SCD [61].
Underlying Disease
Coronary Artery Disease and Acute Myocardial Infarction
As discussed earlier, CAD is the most common cause of SCD in Western countries. Approximately 80 % of patients who experience SCD have a history of CAD. Death in this population may occur in the acute ischemia phase or long after a previous MI. In an autopsy-based study of 902 cases of adjudicated unanticipated sudden cardiac death, 79.3 % of SCD victims had underlying cardiac pathology. Coronary artery disease was the cause of 73.2 % of SCD among persons 35 or older and 23.2 % in those under 35 [62].
Coronary artery disease with more than 75 % cross-sectional stenosis is found in 40–86 % of SCD survivors, depending on the age and sex of the study population [2]. Autopsy studies have reported that a recent occlusive coronary thrombus was found in 15–64 % of victims of SCD caused by ischemic heart disease. A study by Roberts and associates [63] found intraluminal thrombi in 29 % of victims of SCD. However, the thrombus was nonocclusive in more than 80 % of this group. In a study of 90 persons who died as a result of SCD, acute MI was present in 21 % and healed MI in 41 %; no MI was observed in 38 % of the examined hearts [64]. Active coronary lesions (plaque rupture or coronary thrombosis) were identified in 57 % of the entire group of sudden coronary death victims. These data suggest that myocardial ischemia is a major cause of SCD in patients with CAD. Evidence of MI on the basis of elevated cardiac enzymes occurs in less than 50 % of patients with VF, and less than 25 % have Q-wave MI [2]. Many factors can play a role in the process of SCD in patients with a history of CAD. The main factors are acute MI, ischemia without infarction, electrical instability, and structural alterations, such as left ventricular dysfunction [34].
Observation during the ambulatory monitoring of victims of SCD with a history of previous MI showed that the most common mode of death was either VF or VT deteriorating to VF. In the prethrombolytic era, the expected mortality during the first 2–5 years after MI was a little greater than 15 % [65], with three quarters of all deaths being arrhythmic and about 70 % of them being witnessed. Studies in the postthrombolytic era have shown that the incidence of cardiac and arrhythmic death after MI have been substantially reduced, with figures of about 5 and 2 %, respectively, at 2.5 years’ follow-up [66, 67]. Data from more recent studies indicate that the incidence of SCD after MI has declined significantly over the past 30 years because of advances in managing acute MI and now is less than 1 % per year among patients who receive standard medical therapy and revascularization [68].
Mapping of ventricular activation during VT [69] showed the presence of fragmented, repetitive, low-voltage electrical activity in the area of abnormal impulse formation covering most of the interval between two successive tachycardia beats. This was initially interpreted as an indication of reentry circuit with a zone of slow conduction [70]. However, it was later shown that although the cells incorporated in the circuit may have a normal intracellular structure with normal electrical properties, they have lost lateral intercellular connections that lead to a long, maze-type circuit, suggesting a marked slowing of conduction velocity in the circuit [71, 72]. Sudden death due to VF is most common in the 6 months after MI. It is independently predicted by LVEF [38, 73], extent of CAD, premature ventricular beats [74], evidence of ischemia during post-MI exercise testing [75], late potentials [76], and decreased heart rate viability [77]. Approximately one-third of SCD survivors were not inducible during EP testing despite aggressive programmed stimulation [78, 79]. This may represent a population in whom ischemia is important as a trigger or to facilitate the induction of reentrant arrhythmias through modulation of the underlying EP substrate [4].
The incidence and importance of bradyarrhythmias as a mechanism of SCD is difficult to assess. Severe bradycardia, asystole, or electromechanical dissociation is generally considered to account for about 25 % of SCD [80]. In patients with advanced heart failure who are scheduled for cardiac transplantation, this percentage may be as high as 62 % [81]. However, these data supporting a bradyarrhythmic origin of SCD were derived primarily from small groups of patients undergoing long-term ECG recordings at the time of death.
Hypertrophic Obstructive Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant cardiac muscle disorder caused by mutations in genes that affect sarcomeric/myofilament, Z-disc, and calcium-handling proteins [82]. It results in small-vessel disease, myocyte and myofibrillar disorganization, and fibrosis with or without myocardial hypertrophy [83]. These features may result in significant cardiac symptoms and are a potential substrate for arrhythmias. The estimated prevalence of HCM is approximately 1 in 500 [37, 83]. The incidence of SCD associated with HCM has been reported to be 2–4 % per year [84–86]. According to more recent studies, the incidence of SCD is almost 1 % per year among HCM patients [87–89]. The authors attributed previous reported excess mortality rates to selection and referral bias [90]. The incidence of SCD is higher in young patients than in elderly patients with HCM. It is the most common cause of sudden death in competitive athletes younger than 35 years of age (Fig. 24.6) [91].
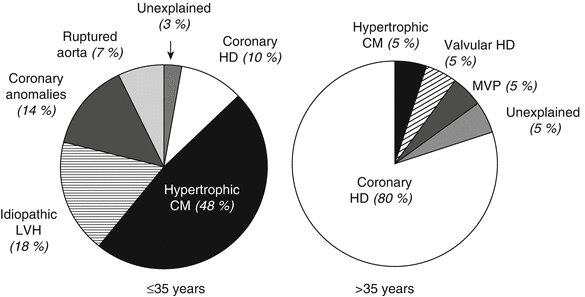
Fig. 24.6
Causes of sudden death in competitive athletes. Estimated prevalences of disease responsible for death are compared between younger (≤35 years) and older (>35 years) athletes. CM cardiomyopathy, HD heart disease, LVH left ventricular hypertrophy, MVP mitral valve prolapse (From Maron et al. [91]. Reprinted with permission from Elsevier Limited)
Since most sudden deaths occur in young asymptomatic or mildly symptomatic individuals, a major focus of managing HCM is identifying people at increased risk for SCD. Despite intense investigation, identifying persons at high risk remains a challenge. The variety and interrelation of mechanisms that can result in SCD represent different aspects of the same phenomenon. Variables that seem to identify patients at increased risk include prior cardiac arrest, spontaneous sustained VT, family history of sudden death, high-risk genotype, multiple repetitive nonsustained VT (NSVT) on ambulatory Holter monitoring or during exercise testing, recurrent syncope, and severe LVH of more than 3 cm [83, 92, 93]. Although the magnitude of the LV outflow gradient was initially considered a risk for sudden death, data have not shown an association [94].
During upright exercise testing, HCM patients commonly show an abnormal blood pressure response, with either a fall or failure of blood pressure to rise. This vascular response is useful in assessing SCD risk predominantly by virtue of a normal test result identifying the low risk young subsets. The absence of an abnormal blood pressure response has a negative predictive value for sudden death, which is 97 % in the young population [83, 95]. This vascular response can be detected in 25 % of HCM patients; thus, its positive predictive accuracy for SCD is low at 15 % [83, 95]. It is a more sensitive indicator of risk in patients younger than 40 years and is associated with sudden death, although the relative risk is low (1.8) [96]. Therefore, a positive result should be used in conjunction with other risk factors.
An angiographic study of children with HCM suggested that myocardial bridging was a significant risk factor for SCD [97]. However, by virtue of having to undergo angiography, these children constituted a highly select group. They were examined retrospectively, making these findings difficult to extrapolate to the general pediatric HCM population. The significance of myocardial bridging and ischemia in initiating secondary arrhythmias remains unknown. However, the available data do not provide sufficient justification for routine angiography.
Other noninvasive electrophysiologic investigations have been used to assess the risk stratification with little success. Furthermore, there is no convincing evidence that EP testing has an important role in identifying patients with hypertrophic cardiomyopathy who are at high risk for sudden death [93].
Some studies [98] have suggested that inducible VT/VF during programmed electrical stimulation in an EP lab in a patient with HCM is associated with a higher risk of cardiac events. However, the response to programmed stimulation is highly dependent on the protocol used. An aggressive protocol using three or more premature stimuli can be expected to produce sustained polymorphic VT in up to 40 % of patients with low predictive accuracy for SCD [99, 100]. Therefore, the hazard and inconvenience of electrophysiologic studies cannot be justified in this population. Available data on genetic markers of SCD in patients with high-risk HCM suggest that β-myosin heavy chain mutations may account for 30–40 % of cases of familial HCM [80, 81, 101]. The prognosis for patients with different myosin mutations varies considerably. Genotype-phenotype correlation studies have shown that mutations carry prognostic significance. Nevertheless, the genotype-phenotype relation has to be clarified further to allow proband risk prediction, as the existing data have been elicited from select groups of patients and their families. β-myosin mutations are heterogeneous in their associated levels of risk. The ARG-403GLN, ARG453CYS, and ARG719TRP mutations in the β-myosin heavy chain are associated with a high incidence of SCD, while the VAL606MET mutation appears to carry a better prognosis.
Troponin-T mutations can be exceptionally lethal and appear to be more homogeneous in their high level of risk than prognostic allelic heterogenicity, which characterizes the other sarcomeric gene abnormalities. Troponin-T patients tend to exhibit a mild degree of hypertrophy [102] but with significant myocyte disarray and therefore may be at high risk of SCD without conspicuous evidence of disease.
All HCM patients should undergo a comprehensive assessment of SCD risk factors (Table 24.1) [103]. Implantable cardioverter-defibrillator implantation is the most effective therapy for HCM patients at high risk of SCD. The 2011 ACCF/AHA guidelines state that an ICD is reasonable in patients with family history of SCD, severe LVH, or unexplained syncope and can be useful in individuals with nonsustained ventricular tachycardia (NSVT) or abnormal systolic blood pressure response to exercise (ABPRE) in the presence of other risk factors. Implantable cardioverter-defibrillators D are not recommended in the absence of risk factors, and the benefit of ICD in HCM patients with only NSVT or ABPRE is uncertain [104, 105].
Table 24.1
Risk factors for SCD in HCM
Established risk markers |
Prior history of ventricular fibrillation, SCD, or sustained VT |
Family history of SCD |
Syncope |
Nonsustained ventricular tachycardia |
Left ventricular wall thickness ≥30 mm |
Abnormal systolic blood pressure response to exercise (failure to increase by at least 20 mmHg or a decrease of ≥20 mmHg) during effort |
Potential SCD risk modifiers |
Left ventricular outflow obstruction |
Late gadolinium enhancement on cardiovascular magnetic resonance imaging |
Left ventricular apical aneurysm |
Genetic mutations |
Coronary Artery Abnormalities
A higher incidence of coronary anomalies has been consistently observed in young victims of sudden death than in adults undergoing routine autopsy (4–15 % in the young vs. 1 % in adults) [106]. Coronary artery anomalies are the second most common underlying pathology associated with SCD among young competitive athletes, accounting for 15–25 % of cases [107]. There is an increased risk of SCD in patients with an anomalous origin of the left main coronary artery from the right (anterior) sinus of Valsalva, with passage of the left main coronary artery between the aorta and the pulmonary trunk. About 75 % of patients reported with this malformation die before age 20, usually during or soon after vigorous exertion [108]. The mirror image coronary anomaly, in which the right coronary artery originates from the left sinus of Valsalva, has also been associated with an increased risk for SCD, although the risk is not as high as the former congenital anomaly [109]. Other unusual variants of coronary artery anomalies, including hypoplasia of the right coronary and the left circumflex arteries [85], the left or right coronary artery originating from the pulmonary trunk, and coronary arterial intussusception that causes coronary lumen occlusion [110], could be rarely associated with SCD. Myocardial bridges have also been associated with SCD during exercise in healthy individuals [111]. It has been suggested that dynamic mechanical obstruction may cause myocardial ischemia. However, evidence that myocardial bridges are responsible for sudden death remains controversial [112]. Coronary dissection with or without aortic dissection occurs in patients with Marfan syndrome [113]. Among other rare mechanical causes of SCD is a rupture of the sinus of Valsalva aneurysm with involvement of the coronary arteries [114]. Prolapse of myxomatous polyps from the aortic valve into coronary arteries has also been reported as a rare cause of SCD [115]. Coronary spasm can cause ischemia and SCD [116]. Vasculitis of coronary arteries, as seen in polyarteritis nodosa, can be associated with SCD [117], and Kawasaki disease can cause SCD through involvement of the coronary arteries [118].
Arrhythmogenic Right Ventricular Dysplasia
Arrhythmogenic right ventricular dysplasia (ARVD) is characterized by ventricular arrhythmias and specific right ventricular cardiomyopathy that shows fatty infiltration on the right ventricle [119]. The term arrhythmogenic right ventricular dysplasia was first proposed in 1977 by Fontaine and colleagues [119] in a report of six patients with sustained VT who were resistant to medical therapy and did not have overt heart disease. In the three patients who underwent surgery, the right ventricle was dilated and had paradoxical wall motion. Arrhythmogenic right ventricular dysplasia should be considered as a possible differential diagnosis in patients with frequent premature ventricular beats or VT, particularly if the ventricular arrhythmias have a left bundle branch block morphology [120]. It is a rare but important cause of sudden death in young, otherwise healthy populations and an insidious cause of congestive heart failure (CHF) [121].
The estimated prevalence of ARVD in the general population is 1:1,000–1:5,000 and typically occurs in young adults. It affects men more frequently than women, with an approximate ratio of 3:1 [122]. The disease is common in northern Italy, where it has a prevalence of 1 in 1,000 [123]. Arrhythmogenic right ventricular dysplasia is an inherited cardiomyopathy with incomplete penetrance typically caused by mutations in genes encoding elements of the cardiac desmosome [124]. It is an autosomal dominant disease in almost 50 % of cases. An autosomal recessive variant of ARVD that is associated with wooly hair and palmoplantar keratoderma has been reported from the Greek island of Naxos [125].
The most common form of presentation includes ventricular arrhythmias, ranging from symptomatic to asymptomatic isolated ventricular extrasystoles to sustained poorly tolerated VT with left bundle branch morphology. Sudden unexpected death could be the first presentation of the disease [126]. Ventricular fibrillation and SCD may be observed during competitive sports and strenuous exercise [127], but they can also occur at rest or even during sleep [128]. The data suggest that, like other inherited cardiomyopathies, it is one of the major causes of SCD in people <35 years old, accounting for up to 25 % of deaths in young athletes [129–131].
The ECG in sinus rhythm shows changes compatible with right-sided abnormality. The first signs to attract medical attention are repolarization abnormalities in terms of T-wave inversion in precordial leads beyond V1 and particularly between V1 and V3, which are observed in 54 % of cases [123]. Extension of T-wave inversion in all precordial leads had a positive correlation with left ventricular involvement [132]. In patients with suspected ARVD, a QRS duration of more than 110 ms in leads V1 to V3 has a sensitivity of 55 % and a specificity of 100 % for this condition [133]. In 25–33 % of ARVD patients, a more specific change with the presence of a discrete just beyond the QRS complex at the beginning of ST segment can be observed during standard ECG evaluation. This deflection has been named the epsilon wave (Fig. 24.7). These waves represent potentials of small amplitude, suggesting delayed ventricular activation of some portion of the right ventricle.
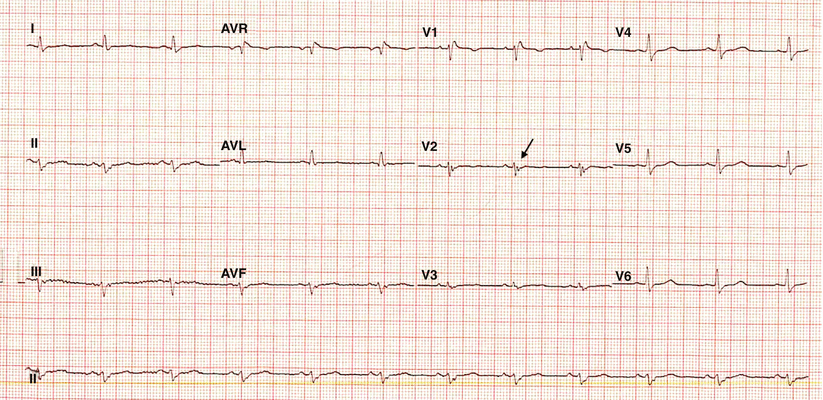
Fig. 24.7
An electrocardiogram (ECG) showing the presence of the epsilon wave (arrow)
Valvular Heart Disease
Aortic stenosis was one of the most common noncoronary causes of SCD in the pre–valvular surgery era. However, the risk of sudden cardiac death [134] is less than 1 % per year in patients with asymptomatic aortic stenosis. Both ventricular arrhythmias and bradyarrhythmias have been associated with SCD in this population. The primary cause of ventricular arrhythmia in this population is believed to be subendocardial ischemia due to LVH and high end-diastolic intracavitary pressure. The arrhythmia may be due to atrioventricular block caused by calcium penetration in the conduction system or neurocardiogenic mechanism. Patients with aortic valve replacement remain at some risk for SCD caused by arrhythmias, prosthetic valve dysfunction, or coexistent CAD [135]. Sudden cardiac death has been reported to be the second most common mode of death after valve replacement surgery, with an incidence of 2–4 % over a follow-up period of 7 years, accounting for 21 % of postoperative deaths. The incidence peaked 3 weeks after surgery and then plateaued after 8 months [136].
It is not clear whether mitral valve prolapse can cause SCD. Its prevalence is so high that its presence may be just a coincidental finding in victims of SCD. Severe mitral regurgitation, LV dysfunction, and myxomatous degeneration of the valve can be markers for patients with higher risk for complications such as endocarditis, cerebral embolic events, and SCD [2]. It has been shown that patients who have valvular heart disease may develop bundle branch reentrant tachycardia, particularly after valvular replacement [137, 138]. The arrhythmia usually occurs in the immediate postoperative period and can result in either cardiac arrest or syncope [138]. Almost all VTs occurred within 4 weeks after surgery (median of 10 days). Because of the proximity of the His-Purkinje system, valvular surgery may result in His-Purkinje system conduction abnormalities that facilitate bundle branch reentry.
Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is the most common type of nonischemic cardiomyopathy, and approximately 10 % of SCDs in the adult population occur in patients with DCM [139, 140]. Overall survivor rates after clinical diagnosis have been reported to be 70 % at 1 year and 50 % at 2 years [141, 142]. The annual mortality in DCM has been reported to range from 10 to 50 %, depending on the severity of disease, with up to 28 % of deaths classified as sudden [2, 142]. More recent studies of the DCM patients on optimal medical therapy have reported considerably lower mortality rates of around 7 % at 2 years [143]. Mortality rates increased with higher New York Heart Association (NYHA) class, but the proportion of patients dying suddenly rather than from progressive pump failure is highest among those with less severe heart failure (NYHA class II or III) [144]. Sudden cardiac death accounts for at least 30 % of all deaths in DCM and may occur in patients with advanced as well as mild disease and in those who appear clinically and echocardiographically to have recovered.
In DCM, malignant ventricular arrhythmias are not the only cause of SCD. Reports vary on the extent to which other mechanisms could be responsible for SCD. In advanced forms of DCM, other causes, such as bradyarrhythmias, systemic embolization, pulmonary emboli, or pulseless electrical activity, may account for up to 50 % of cardiac arrests [142, 145]. However, malignant ventricular arrhythmia is the single most common cause of SCD in DCM. Predictors of overall mortality include EF, end-diastolic dimension or volumes, male sex, old age, hyponatremia, persistent third heart sound, sinus tachycardia, elevated pulmonary/capillary wedge pressure, systemic hypertension, and atrial fibrillation [146].
In a study of noninvasive arrhythmia risk stratification in idiopathic dilated cardiomyopathy, Grimm and associates [147] concluded that reduced LVEF and lack of beta-blocker use were important arrhythmia risk predictors in idiopathic DCM, whereas signal averaged ECG, baroreflex sensitivity, heart rate variability, and T-wave alternans do not seem to be helpful for arrhythmia risk stratification. The major shortcoming of EF and other variables that reflect disease severity is the lack of specificity for arrhythmic death.
Other investigators have focused on syncope and ventricular arrhythmia. In a study by Middlekauff and associates [148], the probability of SCD was 45 % among patients with NYHA functional class III to IV who had unexplained syncope in 1 year. This risk factor was specific for SCD and did not predict the risk of dying from progressive heart failure. Nonsustained VT correlates with disease severity and is seen during ECG monitoring in approximately 20 % of asymptomatic or mildly symptomatic patients and in up to 70 % of severely symptomatic patients [149, 150]. Kron and colleagues reported that NSVT was a sensitive (80 %) but not specific (31 %) marker of SCD [149]. A significant association between the presence of couplets, NSVT, or PVCs of more than 1,000 per day and SCD was reported in a study of 74 patients with dilated cardiomyopathy, NYHA class II to III, 12 of who died suddenly [150]. Inducibility of VT during programmed electrical stimulation predicts sudden death [151], but failure to induce VT does not guarantee that the patient will not have an SCD [149]. In a meta-analysis of 6 programmed electrical stimulation (PES) studies, which included a total of 288 DCM patients, PES failed to identify 75 % of patients who died suddenly [152]. The role of microwave T-wave alternans for arrhythmia risk stratification in DCM patients has to be determined. Several studies evaluated its role for risk stratification of SCD. Klingenheben et al. [153] observed 13 arrhythmic events in 107 patients with CHF of mixed pathogenesis, including 40 patients with nonischemic DCM. Of the 13 patients with arrhythmic events during follow-up in this study, 11 had positive T-wave alternans, and 2 had indeterminate T-wave alternans results, although a negative T-wave alternans test predicted freedom of arrhythmic event in all patients. Kitamura et al. [154] investigated 104 patients with DCM and observed major arrhythmic events in 12 of 83 patients (14 %) during a mean follow-up of 21 months after 21 patients with an indeterminate T-wave alternans test had been excluded from the analysis. As a result, Kitamura et al. found that 11 of 12 arrhythmic events occurred in patients with a positive T-wave alternans test, with additional arrhythmia risk for patients with an onset heart rate of T-wave alternans of ≤100 beats per minute (bpm). Finally, Hohnloser et al. [155] found that a positive microvolt T-wave alternans analysis was the only significant arrhythmia risk predictor by multivariate analysis in 137 patients with DCM during mean follow-up of 14 months. In contrast to these studies, the study by Grimm et al. [147] did not find a positive T-wave alternans to be associated with an increased arrhythmia risk in this population, whereas a negative T-wave alternans test showed a trend toward decreased arrhythmia risk by univariate analysis but not by multivariate analysis.
Pathophysiology of Sudden Cardiac Death
While the common final pathway for SCD is malignant ventricular arrhythmia, the cascade of events that lead to such an end point is variable and depends on the underlying structural, electrical, or environmental abnormalities. The most relevant of these are discussed in the following subsections.
Stable Coronary Artery Disease
Stable CAD is the leading cause of SCD, even in patient populations that did not have a premorbid diagnosis of CAD [156, 157]. A healed MI has been found in up to 70 % of patients with SCD [158]. The incidence of SCD after MI has decreased during last 30 years and now is less than 1 % per year among patients who receive optimal medical therapy and revascularization [4]. Although there is a strong association between CAD and SCD [159], the role of ischemia in SCD is less clear. The results of pathologic studies of victims of SCD have been mixed [160, 161], with some studies showing that as few as 20 % of victims have acute coronary thrombosis and others showing an incidence greater than 95 %. Stable plaques and chronic changes were observed among 50 % of SCD patients with CAD in autopsy-based studies [64, 162, 163].
Evidence does not suggest a clinical progression of ischemic symptoms before SCD [164], and it has been uncommon for monitored patients to show ischemic changes before VF [165, 166]. Ischemia leads to a cascade of events that include calcium overload, decrease in pH, and inhibition of the sodium-potassium pump, all of which may then lead to triggered activity in the form of delayed afterdepolarizations [167, 168]. Delayed afterdepolarizations are associated with any state of calcium overload and therefore can trigger VF [169].
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is the most common cause of SCD in young athletes [125]. The incidence of SCD in people with HCM is approximately 1 % per year [89]. Most people who die from SCD caused by HCM had no symptoms before death. Deaths caused by HCM usually occur during start-stop sports like soccer and football and rarely occur during endurance events, such as long-distance cycling and running [171]. Although there is a correlation between the degree of hypertrophy and the incidence of SCD, the actual mechanism leading to SCD has not been clearly established. There is no clear association between diminished LV filling, LV noncompliance, or outflow tract gradients and SCD [172, 173]. Patients with SCD and HCM have a higher incidence of myofibrillar disarray than patients with HCM who die of heart failure [174]. It is thought that such disarray increases anisotropic conduction and acts as the substrate for microreentrant circuits and VF [175]. Recent studies suggested that ventricular tachyarrhythmia and/or VT is the most probable mechanism of SCD in HCM [176, 177].
Dilated Cardiomyopathy
Patients with CHF have up to a ninefold increase in the risk of SCD compared to the general population [178]. Prolongation of the action potential duration (APD) is a uniform finding in failing hearts [179]. This is in part due to the down regulation in potassium channels, which leads to APD prolongation by delaying the repolarization phase of the action potential (AP) [180, 181]. Furthermore, this change in APD is not uniform throughout the myocardium [182]. The role of APD prolongation in the onset of VF has not been clearly defined, but the extent to which dispersion of repolarization accompanies such changes may be a determining factor [53].
Changes in calcium homeostasis may also play a role in SCD [53]. The calcium channels show a slower rate of decay [183], and the calcium transient’s amplitude is decreased [184]. However, these changes are also nonuniform [185], thus raising the possibility that further perturbations in AP dynamics are created [186].
Slowed conduction and poor electrical coupling have been shown consistently in patients with CHF [187]. These factors facilitate reentry and increase the likelihood of SCD. Conduction velocity is associated with sodium channel current, and the levels of these currents have been found to be attenuated in heart failure [188, 189]. Fibrosis leads to the same findings and is associated with a decreased safety factor for propagation [190]. If conduction fails (blocks), one of the criteria for initiation of reentry has been satisfied. This also predisposes the heart to reentrant ventricular arrhythmias.
Finally, neurohumoral factors may play a role in the onset of ventricular arrhythmias [191]. Activation of the renin-angiotensin-aldosterone system is well documented in these patients, and the blockade of these systems has been shown to decrease overall mortality and SCD [192, 193]. The failing heart has heterogeneities of sympathetic innervation that are thought to be Arrhythmogenic [194, 195].
Dysrhythmias During Sudden Cardiac Death
Ventricular fibrillation is the most common cause of SCD. It is the first rhythm documented in approximately 75 % of patients with cardiac arrest [196]. However, it is most likely that the actual dysrhythmia originates as ventricular tachycardia that subsequently degenerates into VF. A study of 157 patients with ambulatory Holter monitors during cardiac arrest found this to be the case in 62 % of patients [197]. Ventricular fibrillation without antecedent VT occurred in only 8 % of cases.
Bradycardia is not thought to be as common an initiating rhythm of SCD as ventricular arrhythmias [198], although it appears to be the presenting dysrhythmia in patients with more advanced heart failure more often [199]. Bradyarrhythmia and asystole account for 10 % of SCD cases [200]. The role of pulseless electrical activity (PEA) or asystole is more prominent in SCD in people older than 65 [201]. Bradyarrhythmia is more often seen with a nonischemic cardiomyopathy, but asystole or PEA is commonly associated with pulmonary embolism [202].
After coronary occlusion, there are two peaks in the acute incidence of ventricular arrhythmias [203]. The first occurs 10 min before occlusion and the second at 15–20 min after occlusion. These are typically polymorphic VT, presumably due to the acute ischemia-induced derangements in membrane depolarization, repolarization, and refractoriness.
Subsequently, VF can occur within the first 4 days after infarction. These are usually initiated by PVCs, the mechanism of which is thought to be abnormal automaticity [204]. The macroreentrant circuits leading to monomorphic VT evolve during chronic stages after the myocardium has healed and scar tissue has developed [64].
The GUSTO-1 trial showed that the overall incidence of VT or VF was 10.2 % (VF: 4.1 %, VT: 3.5 %, both VT and VF: 2.7 %) among patients with acute ST elevation MI and approximately 80 % of them occurred within 2 days after acute MI [205]. In another study, the overall incidence of VT or VF was reported as 2.1 % (VF: 1 %, VT: 0.8 %, both VT and VF: 0.3 %) after non-ST elevation acute coronary syndrome. The median time of arrhythmia occurrence was 78 h [206].
Evaluation and Risk Stratification
Although multiple diagnostic tests have been used to evaluate different cardiac and noncardiac factors that play a role in the occurrence of SCD during the past 25 years, the relatively low positive predictive accuracy of these tests affects their usefulness [207]. Our understanding of these risk factors is incomplete. When screening a patient for the presence of risk factors for SCD, one should first evaluate the underlying cardiac pathology and the presence of possible comorbid noncardiac conditions. The first step is taking a complete history and physical examination, which can provide clues about the patient’s risk of SCD. Because CAD is the most common underlying factor in SCD, particular attention should be directed toward a history of chest discomfort or recent exertional intolerance. Because LV dysfunction is a major risk factor for SCD, potential symptoms of CHF should be carefully evaluated. A history of cardiac arrest is the most significant risk factor for recurrent cardiac arrest [208]. In patients with structural heart disease, particular attention should be paid to history of an unexplained syncope, which puts this population at higher risk for SCD [207]. In unexplained syncope in patients with structural heart disease or in patients who survive SCD, interviewing those who witnessed the event can provide crucial information. Documentation of all rhythm strips recorded during the event is also paramount. Any current use of cardiac or noncardiac drugs or prescribed or over-the-counter medications must be carefully determined because of the possibility of QT prolongation.
The patient’s medical history should include any family history of hypertrophic cardiomyopathy, Marfan syndrome, and sudden or unexplained death. A careful physical examination also provides further insight into the presence of underlying structural heart disease and all other comorbid conditions. Various noninvasive methods are used to evaluate the underlying cardiac pathology and help the risk stratification process.
Electrocardiography
An ECG is helpful in diagnosing underlying CAD and MI. Furthermore, it provides other helpful markers, such as QT interval (prolonged QT interval in acquired and congenital LQTS, short QT interval in short QT syndrome), delta wave (a clue to Wolf-Parkinson-White syndrome), epsilon wave (in ARVD), and right bundle branch block and ST segment elevation in V1 to V3 (in Brugada syndrome). An ECG is also a specific but insensitive tool with which to evaluate LVH.
Echocardiography
Echocardiography provides information regarding LVEF, one of the most powerful predictors of recurrent cardiac arrest [209]. Left ventricular ejection fraction is an independent predictor of death. An LVEF ≤0.40 indicates an increased risk of death by at least three- to fourfold [210].
However, when the LVEF is severely depressed (≤15–20 %), the prevailing mode of cardiac death is not sudden. When it is sudden, it is often related to bradyarrhythmias or electromechanical dissociation rather than ventricular tachyarrhythmias. The meta-analysis of pooled data from the European Myocardial Infarction Amiodarone Trial (EMIAT), the Canadian Amiodarone Myocardial Infarction Trial (CAMIAT), Survival with Oral d-Sotalol (SWORD), Trandolapril Cardiac Evaluation (TRACE), and the Danish Investigations of Arrhythmia and Mortality on Dofetilide study group (DIAMOND) assessed the risk of death in patients who survived at least 45 days after MI [211]. The prognostic value of EF was adjusted for treatment and other demographic factors associated with survival. The meta-analysis confirmed that LVEF significantly predicted 2-year, all-cause arrhythmic and cardiac mortality. A 10 % absolute increase in EF reduced the mortality at 2 years with a hazard ratio of 0.61. The EF is usually combined with other risk factors. While it is unclear which combination of noninvasive variables provides the strongest risk prediction in the current thrombolytic era, combining the variables that reflect different factors linked to SCD is logical. These factors could include the substrate (EF), the trigger (ventricular premature beats, NSVT), or the modulator (autonomic dysfunction).
As discussed earlier, the ATRAMI investigators showed that a combination of low values of autonomic markers and reduced EF identified a group of post-MI patients at highest risk for sudden death. The results from another study [212] confirmed prethrombolytic era findings [213] that echocardiographic LV end-systolic and end-diastolic volumes were strong predictors of mortality at 6 months after acute MI. Echocardiography can also be used to evaluate segmental wall motion abnormalities associated with CAD, significant valvular dysfunction, evidence of hypertrophic cardiomyopathy, pericardial disease, intracardiac tumors, and congenital heart disease. The presence of LV dysfunction precludes the use of certain antiarrhythmic agents that can produce a negative inotropic effect or a proarrhythmic event.
Exercise Stress Testing
Exercise stress testing is a recognized prognostic test in survivors of acute MI. Several studies have shown that the presence of ST segment changes, the occurrence of exercise-induced angina, inappropriate blood pressure response, and exercise-induced ventricular arrhythmia in post-MI patients during submaximal predischarge stress testing are predictors of recurrent ischemic events, the need for revascularization, and overall cardiac mortality rates. Several authors have reported that these findings are predictive of ventricular arrhythmia and sudden death [213–216]. However, other investigators have not found exercise test results to specifically predict the risk of SCD [75, 217, 218]. Exercise testing also helps identify patients with exercise-induced or exercise-aggravated ventricular tachycardia [219–221]. Stress testing is most commonly used as a noninvasive test to evaluate the presence of CAD in patients with chest pain. The sensitivity and the specificity of stress tests improve when the tests are combined with nuclear methods.
Radionuclide Imaging
Radionuclide angiography is another noninvasive method that can be used to quantitatively assess LV function. It can also provide information regarding regional LV performance and myocardial viability.
Magnetic Resonance Imaging
Magnetic resonance imaging can provide information about myocardial viability, LV function, and LV end-diastolic and end-systolic volumes. Furthermore, as discussed earlier, MRI can be a useful tool for evaluating patients with suspected ARVD.
Ambulatory Electrocardiographic Monitoring
The role of Holter monitoring in the evaluation of patients with arrhythmias has been the subject of multiple studies, particularly those of MI patients. Several studies confirmed the prognostic significance of frequent premature ventricular complexes and NSVT in post-MI patients [222, 223]. However, the specificity of a spontaneous ventricular ectopy is limited [224].
Mortality rates are not influenced by the frequency, duration, or rate of NSVT [222, 223]. However, all of the above-mentioned studies were performed in the prethrombolytic era. In the thrombolytic era, the risk associated with the presence of NSVT is currently unknown. The GISSI-II study investigators reported that the prevalence of NSVT was only 6.8 % and its presence was not predictive of SCD at 6 months after MI [225]. In another study, there was a low prevalence (9 %) of NSVT shortly after acute MI. At multivariate analysis, NSVT was not an independent predictor of SCD, unlike heart rate variability, EF, or status of the infarct artery. More recent data from primary prevention of SCD by prophylactic ICD implantation have shown that the combination of NSVT with other variables, including reduced EF and electrophysiologic testing after acute MI, was effective in identifying post-MI patients at high risk of arrhythmic death.
Signal-Averaged Electrocardiography
Low-amplitude, fragmented, and delayed electrical activity can be recorded from areas bordering the infarction in an experimental model of MI. The signal-averaged ECG (SAECG) records this delayed fractionated activity from the body surface. A number of studies have evaluated the prognostic significance of this SAECG alone or in combination with Holter monitoring or LVEF in the post-MI population [66, 76, 226, 227]. In these studies, the sensitivity during a follow-up period of 6–24 months in patients who experienced sustained VT or SCD was between 50 and 90 %. Its primary benefit was its excellent negative predictive value, reported to be about 95 %. However, the positive predictive value of SAECG (the risk of arrhythmia in a patient with positive results) has been lower, averaging 20 % in these studies.
Kuchar and colleagues [227] risk-stratified patients after an acute MI by using SAECG, Holter monitoring, and radionuclide ventriculography. The patients were followed for a median of 14 months for an arrhythmic event, defined as sudden death or sustained VT. The results of each of these three tests were independently predictive of arrhythmic events. An LVEF <40 % was the most powerful predictor of an arrhythmic event. Adding a positive SAECG to the LVEF further increased the probability of predicting an event (from 4 % for LVEF of <40 % alone to 34 % with LVEF <40 % plus a positive SAECG). An SAECG is more predictive of arrhythmic events in inferior infarction and is less useful in anterior infarctions. This difference is probably due to the fact that the peri-infarct tissue in anterior infarction is activated relatively early in the sequence of ventricular activation, which makes detecting late potentials difficult.
A meta-analysis of all available prospective studies during the prethrombolytic era on the use of SAECG after MI showed that the SAECG predicted a sixfold increase in risk of arrhythmic events independent of LV function and an eightfold increase in risk of arrhythmic events independent of Holter results [228]. Beta blockers and successful thrombolysis/revascularization reduce the frequency of late potentials after acute MI and significantly diminish the prognostic power of SAECG [229, 230]. Some studies supported the concept that SAECG is an independent predictor of arrhythmic events after MI [230, 231]. The CARISMA study showed that a filtered QRS duration in the SAECG ≥120 ms predicted VF or symptomatic sustained VT after acute MI [232].
Heart Rate Viability
Heart rate viability (HRV) is a measure of beat-to-beat variation of sinus-initiated RR intervals. It has been evaluated as an indicator of decreased parasympathetic tone, which is associated with poor prognosis in post-MI patients. Schneider and Costiloe [233] evaluated the relationship between sinus arrhythmia and prognosis after MI and concluded that sinus arrhythmia decreases in normal patients with age, that sinus arrhythmia is less evident after MI, and that patients with the least evidence of sinus arrhythmia had the worst prognosis during follow-up.
Kleiger and colleagues [234] examined the relationship between increased mortality rates and decreased heart rate viability in a study of 808 post-MI patients. They showed that HRV has a significant relation with other prognostic indicators, relating directly to LVEF and exercise capacity. However, HRV correlated to a much lower degree with ventricular ectopy, suggesting that these two factors acted independently. Farrell and associates [235] observed that the sensitivity of HRV in predicting arrhythmic events (sudden death and sustained VT) was higher than that of other risk factors, including exercise testing, LVEF, ventricular ectopy, and SAECG. In the analysis of a combination of risk factors, the combination of decreased HRV and the presence of late potentials in SAECG was more predictive of arrhythmic events than other combinations. The decrease in HRV suggests a relative decrease in parasympathetic tone [236]. Another possible explanation is that increased vagal tone protects against VF in the presence of ischemia.
As we discussed earlier, the ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) investigators [60] studied HRV and baroreceptor sensitivity on 1,284 patients in the first month after MI. They concluded that these variables were significant predictors of cardiac mortality. They showed that, during 21 months of follow-up, depressed HRV and baroreceptor sensitivity carried a significant multivariate risk of cardiac mortality of 3.2 and 2.8, respectively. The combination of low HRV and depressed baroreceptor sensitivity further increased the risk. One-year mortality increased from 1 % when both markers were well preserved to 15 % when both were depressed. Furthermore, the investigators showed that the predictive power of baroreceptor sensitivity declined much more markedly than HRV in patients older than 65. The ATRAMI investigators have shown that after MI, the analysis of the autonomic markers has significant prognostic value independent of established clinical predictors, such as EF and ventricular arrhythmias. Furthermore, the combination of low values of autonomic markers and reduced EF identifies a group of post-MI patients at high risk for SCD.
Huikuri and his team [237] showed that HRV and heart rate turbulence increased over time after MI in the CARISMA and REFINE studies and attenuated recovery of autonomic function after MI was associated with a 9.4-fold higher risk of ECG-documented sustained VT or VF in CARISMA and a 7.0-fold higher risk of fatal or near-fatal events in REFINE.
Cardiac Catheterization
Cardiac catheterization should be performed in almost all survivors of SCD to establish the presence, extent, and severity of CAD. It can also exclude congenital coronary vessel anomalies in younger SCD survivors. Cardiac catheterization can confirm the results of noninvasive studies for evaluation of LV function, wall motion abnormalities, and valvular disease.
Electrophysiologic Testing
Wellens and colleagues [238] showed that programmed stimulation could safely and reproducibly initiate VT in most patients who experienced sustained VT. Subsequent studies confirmed this observation. Between 60 and 90 % of patients who survive sudden death unassociated with acute MI are inducible during EP study [225, 238–240]. Sustained monomorphic VT can be induced during EP study in 50–60 % of cardiac arrest survivors, and polymorphic VT or VF can be induced in an additional 10–20 % [241–243]. However, in the thrombolytic era, EP testing for risk stratification of patients has progressively lost favor. Nearly half of all reported trials found inducibility of sustained VT during programmed stimulation unable to predict later mortality or arrhythmic events [244]. Many post-MI patients with SCD have negative predischarge electrophysiologic tests, resulting in a low negative predictive accuracy [245]. Furthermore, when used alone, EF is superior to EP testing in predicting arrhythmic events after acute MI [246]. Therefore, a 2-step strategy using EF ≤40 % and ventricular arrhythmias on Holter monitoring and then electrophysiologic testing significantly improved the positive predictive accuracy of risk stratification process but only to a moderate level of 18.2 % [247].
Moreover, evidence from primary prevention trials (Multicenter Automatic Defibrillator Implantation Trial [MADIT] and Multicenter UnSustained Tachycardia Trial [MUSTT]) confirmed that a 2-step risk stratification procedure using reduced EF and nonsustained VT followed by EP testing was helpful in selecting a high-risk subgroup of patients who benefited from prophylactic ICD implantation for the primary prevention of SCD. However, the precise value of VT inducibility is uncertain. Data from the MADIT-II trial showed that EP testing had moderate predictive value for the occurrence of monomorphic VT, but incidence of VT and VF were not significantly different between inducible and noninducible patients in 2-year post-MI follow-up [248].
Microvolt T-Wave Alternans
Microvolt T-wave alternans (TWA) is beat-to-beat variability in the T wave [249]. Its precise mechanism remains unclear, but one proposed mechanism is beat-to-beat changes in the intracellular levels of Ca2+. The beat-to-beat variability in intracellular Ca2+ leads to modulation of repolarization currents, which may contribute to TWA [250]. Metabolic disturbances during ischemia lead to microvolt TWA [251]. T-wave alternans is thought to contribute to induction of malignant arrhythmias by leading to dispersion of refractoriness [252]. This in turn can lead to functional block and induction of reentrant arrhythmias.
Using the spectral method and sophisticated signal-processing techniques, microvolt TWA is analyzed during standard stress exercise protocols, treadmill stress echocardiography, pharmacologic stress testing, or atrial pacing in the electrophysiology lab [253]. A positive test is the presence of significant sustained alternans measured in any three orthogonal leads or two adjacent precordial leads. It should be present for at least 1 min with an onset at a HR >110 bpm [253]. Microvolt TWA testing is a reliable marker for late post-MI risk stratification, but there is some controversy about its accuracy in patients with recent infarction [254–258]. In patients with idiopathic dilated cardiomyopathy, it has shown promise as a clinically important risk stratifier [253]. Despite promising results in previous studies, data from more recent studies failed to prove reliability of microvolt TWA as a predictor of serious arrhythmic events [259]. There are only limited data for microvolt TWA testing in patients with hypertrophic cardiomyopathy or the inherited arrhythmic disorders.
Prevention
The majority of patients who have an SCD do not have symptoms and are not identified as being high risk before the event [248]. Therefore, in addition to the secondary prevention of SCD (prevention of recurrent cardiac arrest), primary prevention is a major therapeutic goal. As discussed earlier, patients with the highest risk factor profile constitute a small percentage of the total number of people at risk for SCD. Furthermore, when the high-risk subgroups are identified and removed from this population base, the calculated incidence for the remainder of the population decreases and the identification of individuals at high risk becomes more difficult. During the past decade, multiple trials have been conducted on the primary prevention of SCD in patients with heart disease who are high risk and the secondary prevention of SCD in patients who have been successfully resuscitated. Here, we summarize pertinent data from the extensive literature, but reviewing extensive data from numerous trials is beyond the scope of this chapter.
Primary Prevention
Pharmacologic Studies
Beta Blocker Therapy
Available data from several prospective double-blind studies revealed that beta blockers reduce the overall mortality and SCD rates after acute MI. In the beta blocker heart attack trial (BHAT) [260], propranolol (180–240 mg per day) decreased the total mortality rate over an average follow-up period of 25 months by 26.5 % (from 9.8 % in the placebo group to 7.2 % in the propranolol group). The benefit was remarkable in high-risk patients. Propranolol reduced the risk of death in this group by 43 % (p < .001) [261]. Propranolol decreased the incidence of SCD by 47 % in patients who had previous heart failure versus 13 % in the patients who did not, with a 35 % reduction in adjusted mortality rate.
In the Norwegian multicenter study on timolol after an acute myocardial infarction [262], timolol reduced the total mortality by 38 %. Sudden cardiac death decreased by 45 % from 13.9 % in the placebo group to 7.7 % in the timolol group (P = .0001). The beneficial results persisted for up to 72 months [263].
In the Acebutolol Postinfarction Trial (Acebutolol et Prevention Secondaire de L’Infarctus, APSI) [264], acebutolol reduced the mortality rates by 48 % and cardiovascular death by 58 % compared with a placebo. The benefit was maintained after several years of follow-up [265]. In the Goteborg trial [266], metoprolol (intravenous infusion followed by 200 mg a day postoperatively) reduced mortality rates by 36 %, from 8.9 to 5.7 %.
In the Metoprolol in Acute Myocardial Infarction (MIAMI study) [267], the metoprolol group had statistically insignificant 13 % reduction in mortality rate. However, retrospective analysis showed that the treatment was beneficial in high-risk patients and reduced mortality rate from 8.5 to 6 % (p = .03). Metoprolol was also used in the Thrombolysis in Myocardial Infarction (TIMI-IIB study) [268] as an adjunct to intravenous tissue plasminogen activator. The patients were randomly assigned to receive immediate intravenous metoprolol followed by oral therapy or to defer therapy with metoprolol starting on day 6 after the MI. There was a lower rate of recurrent ischemia and nonfatal MI in the group that received immediate therapy.
In a meta-analysis of 26 trials by Yusuf and associates [269], therapy with beta-blockers resulted in a 23 % reduction in mortality rates. The mechanism of beneficial effects of beta blockers is unclear. The survival benefits appear to result from a reduction in arrhythmic-related death and recurrent MI. Beta blockers reduce the threshold for VF, most likely through the antisympathetic effect. Beta blockers also reduce hyperkalemia by blocking the catecholamine-induced influx of potassium into cells [270]. Patients with depressed LV function and a history of CHF show the greatest survival benefit. Beta blockers may improve survival in patients with CHF by reducing myocardial oxygen demand, improving diagnostic relaxation, reducing sympathetic mediated vasoconstriction and tachycardia, or reducing catecholamine-induced myocardial damage or by their antiarrhythmic effect [270].
Beta blockers are also effective for managing idiopathic right ventricular outflow tract VT and catecholaminergic polymorphic VT, which are rare causes of SCD [271]. Bisoprolol was noted to be beneficial in the Cardiac Insufficiency Bisoprolol (CIBIS) study [272]. Although there was no overall benefit in survival, there was a 57 % reduction in mortality rate in patients with previous MI. In the CIBIS-II study [273], bisoprolol showed a significant mortality benefit. In this multicenter, double-blinded, randomized, placebo-controlled trial, all controlled patients were in NYHA functional classes III to IV with an LVEF of <0.35. The all-cause mortality rate was significantly lower with bisoprolol (11.8 % vs. 17.3 % in the placebo group, p < .0001). Furthermore, the incidence of SCD decreased by 44 % among patients receiving bisoprolol (3.6 % vs. 6.3 % in placebo group, p = .0011).
In the mortality effect of metoprolol in patients with heart failure (MERIT-HF trial) [143, 274], treatment with metoprolol CR/XL was associated with a 34 % decrease in all-cause mortality rates, a 38 % decrease in cardiovascular mortality rates, a 41 % decrease in SCD, and a 49 % decrease in death due to progressive heart failure. Carvedilol, a nonselective β-receptor agonist with some α1-receptor antagonist activity, improved survival rates in patients with CHF in a U.S. multicenter study [275]. There was a 65 % reduction in mortality rates. The result of this trial was supported by another trial from Australia and New Zealand [276], which showed a 26 % decrease in death or hospital admission in patients with ischemic cardiomyopathy.
In a more recent study, the CAPRICORN investigators [277] evaluated 1,959 patients with a proven acute MI and LVEF ≤40 % in a multicenter, randomized, placebo-controlled trial. The patients were randomly assigned to carvedilol or a placebo. Carvedilol reduced the frequency of all-cause mortality in the study. The authors reported a 23 % relative reduction in mortality. The reduction in all-cause mortality was in addition to the effects of angiotensin-converting enzyme (ACE) inhibitors and reperfusion therapy, which were prescribed in 98 and 46 % of patients, respectively.
In a multicenter, double-blind, and randomized parallel group trial, the Carvedilol Or Metoprolol European Trial (COMET) [278], investigators assigned 1,511 patients with chronic heart failure to treatment with carvedilol and 1,518 patients to metoprolol. Patients were required to have NYHA class II to IV, previous admission for a cardiovascular reason, an EF <0.35, and to have been treated optimally with diuretics and ACE inhibitors unless not tolerated. The all-cause mortality was 34 % for carvedilol and 40 % for metoprolol (hazard ratio of 0.83, p = .122). The authors concluded that carvedilol extends survival compared with metoprolol in patients optimally treated with diuretics and ACE inhibitors. The absolute reduction in mortality over 5 years was 5.7 %. The COMET trial highlights the need for improved understanding of specific mechanisms of action of selective and nonselective beta blockers in HF patients.
Angiotensin-Converting Enzyme Inhibitors and Angiotensin-II Receptor Antagonist
The beneficial effect of ACE inhibitors appears to be a class effect mediated by a reduction in ventricular size, reinfarction, the appearance of CHF, and a new ischemic event. Several studies have shown a 6–22 % reduction in mortality rate. Despite the beneficial effect on total mortality rate, the precise role of these agents in reducing SCD is still not clear. In the Veterans Administration Cooperative II study (V-HeFT-II) [279], there was a 28 % reduction in mortality rate in the enalapril group from reduced incidence of SCD compared with the hydralazine-isosorbide group. In the Trandolapril Cardiac Evaluation (TRACE) study [280], trandolapril reduced the mortality rates by 22 % and SCD rates by 24 % (p = .03) in post-MI patients with evidence of LV dysfunction. However, other studies did not show any significant reduction in the SCD rate.
In the Survival and Ventricular Enlargement (SAVE) trial [281], although captopril reduced total mortality rates by 19 % in survivors of MI with asymptomatic left ventricular dysfunction (LVEF ≤40 %), there was no statistical difference in the SCD rate. In the Studies on Left Ventricular Dysfunction (SOLVD) prevention trial [282], captopril did not significantly reduce total mortality and SCD rates in asymptomatic (NYHA functional class I to II) patients with an LVEF ≤0.35. More recently, the Heart Outcomes Prevention Evaluation (HOPE) study [283] concluded that treatment with ramipril reduced the rates of death from cardiovascular causes (6.1 % compared to 8.1 % in the placebo group with p < .001), death from any cause (10.4 % vs. 12.12 %, p = .005), and cardiac arrest (p = 0.03). The authors concluded that ramipril, an ACE inhibitor, is beneficial in a broad range of patients who are at high risk for cardiovascular events without evidence of left ventricular systolic dysfunction or heart failure.
The angiotensin-II receptor antagonists that block the receptor without increasing bradykinin levels have the potential to be as effective as or more effective than ACE inhibitors in treating patients with heart failure and possibly reducing the risk of SCD. Angiotensin II can be produced through alternate pathways, which may be an advantage over ACE inhibitors. In the Evaluation of Losartan in the Elderly (ELITE) trial [284], a prospective, randomized, double-blind clinical trial comparing the safety and efficacy of losartan and captopril in patients with documented LVEF <40 %, the mortality rate was 46 % lower in the losartan group than in the captopril group. To further study the results of this trial, the ELITE-II trial evaluated the effects of losartan and captopril on mortality and morbidity in a larger number of patients with heart failure [285]. ELITE-II was a double-blind, randomized, controlled trial of 3,152 patients 60 years or older with NYHA class II to IV heart failure and an EF ≤40 %, who were randomly assigned to losartan or captopril. The primary and secondary end points were all-cause mortality and sudden death or resuscitated arrest. There were no significant differences in all-cause mortality or sudden death or resuscitated arrests between the two treatment groups. In contrast to ELITE-I, the results of ELITE-II suggested that losartan was not superior to captopril in improving survival in elderly patients with HF, but losartan was tolerated significantly better. It was suggested that ACE inhibitors should be the initial treatment of heart failure, although angiotensin-II receptor antagonists may be useful to block the renin-angiotensin system when ACE inhibitors are not tolerated.
The Optimal Trial in Myocardial Infarction with Angiotensin-II Antagonist Losartan (OPTIMAAL) [286] was a multicenter, randomized trial to test the hypothesis that the angiotensin-II antagonist losartan would be superior or comparable to the ACE inhibitor captopril in decreasing all-cause mortality in high-risk patients after acute MI. There was a nonsignificant difference in total mortality in favor of captopril; however, losartan was significantly better tolerated than captopril, with fewer patients discontinuing the study medication. Thus, ACE inhibitors were recommended as a first choice of therapy in patients after complicated acute MI. Evidence suggests that some benefits of ACE inhibitors are derived from elevated levels of bradykinin [287]. Therefore, the combination of an ACE inhibitor and angiotensin-II receptor antagonist may have additive actions.
Several studies evaluated this hypothesis. The Valsartan in Acute Myocardial Infarction Trial (VALIANT) [288] compared the effects of valsartan, captopril, and the combination of valsartan and captopril in a population of high-risk patients with clinical or radiologic evidence of HF, evidence of LV systolic dysfunction, or both after an acute MI. The primary end point of this study was death from any cause. The results of this study showed that there were no additional benefits of using combination therapy in CHF after acute MI over ACE inhibitor alone or with an angiotensin receptor antagonist. Furthermore, the two agents were equivalent in terms of overall mortality and in terms of rate of composite end points of fatal and nonfatal cardiovascular events. Adverse events were less common with monotherapy than with combination therapy. These results were challenged in a much larger trial, the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) [289], a randomized, double-blind, placebo-controlled, clinical trial that compared candesartan with a placebo in patients with symptomatic heart failure. The primary outcome of this study was all-cause mortality. The patients eligible for CHARM were enrolled in three different subgroups according to LVEF: greater than 40 % (CHARM-preserved) [290], 40 % or less and being treated with an ACE inhibitor (CHARM-added) [291], or 40 % or less and not being treated with an ACE inhibitor because of previous intolerance (CHARM-alternative) [292].
Review of the data from the CHARM-added portion of the trial showed that the number of deaths from any cause in the candesartan group was 377 (30 %) compared with 412 (32 %) in the placebo group. These data indicated a trend toward decreased mortality with combination therapy.
However, the Valsartan Heart Failure Trial (Val-HeFT) [293], a randomized, placebo-controlled, double-blind, parallel group trial in patients with NYHA class II to IV, showed that the overall mortality was similar in the two groups. The patients were divided into four subgroups based on use or nonuse of ACE inhibitor and beta-blocker therapy at baseline. In all subgroups except the subgroup of patients treated with an ACE inhibitor and a beta blocker at baseline, valsartan had a significantly favorable effect on the rate of the combined end point. The three groups also had a favorable point estimate of the odds ratio for death.
Aldosterone Antagonists
Spironolactone (the RALES study) [294] achieved a significant 30 % reduction in overall mortality and a 35 % reduction in hospitalization in 1,663 patients with NYHA class III to IV heart failure. In this study, all patients were on ACE inhibitors, but only 10 % were on beta blockers. Using eplerenone in patients with recent MI and LVEF <40 %, investigators in the EPHESUS trial [295] showed a significantly lower all-cause mortality (14.4 % vs. 16.7 %). In this study, 75 % of patients were on beta blockers, and 87 % were on ACE inhibitors. Eplerenone reduced SCD risk by 33 % in patients with baseline LVEF ≤30 % [296]. In the CHARM trial, 17 % of patients were on spironolactone, and a subset analysis in this group did not find significant improvement of the primary end point by the addition of candesartan. Combining the aldosterone antagonist and the angiotensin-II receptor antagonist may not further reduce end points because they both blockade the local renin-angiotensin system in the cardiac muscle and the vasculature via the same pathway [297].
Antiarrhythmic Drug Therapy
Class I Antiarrhythmic Drugs
Frequent and complex ventricular activities in survivors of MI are risk markers for subsequent SCD. The objective of the Cardiac Arrhythmia Suppression Trial (CAST) I and II [298, 299] was to test the hypothesis that the suppression of ventricular ectopy after an MI would reduce the incidence of SCD. In the CAST-I study [298], patients who had asymptomatic PVCs after MI suppressed by encainide or flecainide were randomly assigned to receive long-term drug therapy or placebo. After an average follow-up of 9.7 months, the total mortality was 7.7 % in the class IC group versus 3 % in the placebo group (relative risk 2.5, p = .0001). Arrhythmic death was more common in the class IC group (4.5 % vs. 1.2 % in the placebo group). The relative risk of death of resuscitative cardiac arrest was 2.38. Further analysis showed that the adverse event rate was highest in patients with the lowest LVEF. An LVEF >0.30 was associated with improved survival rates.
In a subgroup analysis, patients treated with beta blockers in addition to class IC antiarrhythmic agents had lower mortality rates than patients treated with class IC agents alone. This suggests a protective effect of beta blockers [299]. The results of the CAST trial disproved the hypothesis that suppression of ventricular arrhythmia improves mortality rates. Furthermore, meta-analysis of other class I agent trials showed a significantly higher mortality rate for antiarrhythmic agent–treated patients than for placebo-treated patients [300].
A third drug, moricizine, was subsequently studied in the CAST-II trial [301]. The overall mortality rate was similar for patients treated with moricizine and those treated with placebo. However, there was a significantly higher mortality rate among patients treated with moricizine during the initial 2 weeks of therapy (2.3 % vs. 0.3 %). The cause of this proarrhythmic response is unknown. The unexpectedly low placebo mortality rate suggests that a low-risk population was chosen for the trial, which exposed the patients to all the risks of active therapy without much hope of benefit.
Class III Antiarrhythmic Drugs
Amiodarone is a unique antiarrhythmic drug with class I, II, III, and IV effects. The effect of amiodarone in preventing SCD was studied extensively in post-MI patients. The Basel Anti Arrhythmic Study of Infarct Survival (BASIS) [302] had a 61 % reduction in mortality rate with amiodarone. Amiodarone also decreased VF or SCD compared with the controlled group (p = .024). The beneficial effect of amiodarone persisted several years after drug discontinuation. The Polish Amiodarone Trial (PAT) [303] showed a reduction in cardiac death rates from 10.7 % in the placebo group to 6.9 % in the amiodarone arm of study.
The goal of the European Myocardial Infarction Amiodarone Trial (EMIAT) [304] was to assess the efficacy of amiodarone in reducing mortality rates in patients with depressed LV function after an MI. This study enrolled 1,486 patients with an LVEF ≤0.40 within 5–21 days after an MI. The median follow-up was 21 months. Patients were randomly assigned to treatment with amiodarone or placebo.
The primary end point was all-cause mortality, and secondary end points were cardiac death, arrhythmic death, and the combination of arrhythmic death and resuscitated cardiac arrest. Amiodarone reduced arrhythmic death by 35 % (p = .05) and arrhythmic death and resuscitated cardiac arrest by 32 % (p = .05). However, amiodarone did not show any beneficial or detrimental effect on all-cause mortality rates.
The Canadian Amiodarone Myocardial Infarction Trial (CAMIAT) [305] evaluated the hypothesis that amiodarone could reduce arrhythmic death among post-MI patients (6–45 days after MI) who had frequent PVCs (≥10 PVCs per hour) or any run of VT on baseline Holter recording. The primary end point was arrhythmic death or resuscitated VF. Secondary end points were arrhythmic death, cardiac death, and all-cause mortality. In the efficacy analysis, resuscitated VF or arrhythmic death occurred in 6 % of patients in the placebo group and 3.3 % of patients in the amiodarone group. Amiodarone reduced the relative risk by 48.5 %. Intention-to-treat analysis showed 38.2 % risk reduction in the amiodarone group compared to the placebo group (6.9 % in the placebo group to 4.5 % in the amiodarone group, p = .029). The absolute risk reduction was greatest among patients with CHF or a history of MI. Although amiodarone reduced all-cause mortality by 18 %, the difference was not statistically significant.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


