CHAPTER
14
Sudden Cardiac Death and Inherited Arrhythmias
SUDDEN CARDIAC DEATH (SCD)
SCD is defined as an unanticipated, non-traumatic death in a stable patient within 1 hour of symptom onset (witnessed) or within 24 hours of being observed alive and symptom-free (unwitnessed).
Anatomy and Physiology (Mechanism)
○ Malignant ventricular arrhythmias (e.g., ventricular fibrillation [VF]) cause 75% of SCDs.
▪ Of these, 45% are ventricular tachycardia (VT) that degenerates to VF.
○ Bradyarrhythmia (heart block, asystole) is the other 25%.
▪ Note: Pulseless electrical arrest (PEA) is increasingly recognized during resuscitation as a causative or contributory rhythm.
Causes of SCD
○Coronary artery disease (CAD; dominant mechanism, ~70%–80%):
▪ Ischemic heart disease or coronary atherosclerosis
▪ Congenital abnormalities of coronary arteries: Anomalous origin, AV fistula
▪ Coronary spasm
▪ Coronary dissection
▪ Coronary artery embolism
▪ Coronary arteritis
▪ Myocardial bridging
○Cardiomyopathies (10%–15%):
▪ Ischemic cardiomyopathy
▪ Hypertrophic cardiomyopathy
▪ Dilated cardiomyopathy
▪ Valvular cardiomyopathy
▪ Alcoholic/toxic cardiomyopathy
▪ Infiltrative (e.g., sarcoidosis, amyloidosis, hemochromatosis, Fabry)
▪ Arrhythmogenic right ventricular cardiomyopathy
▪ Takotsubo cardiomyopathy
▪ Left ventricular non-compaction cardiomyopathy
▪ Myocarditis (e.g., acute, giant cell, chronic lymphocytic)
▪ Neuromuscular diseases (e.g., muscular dystrophy, Friedreich’s ataxia, myotonic dystrophy)
▪ Congenital cardiomyopathy (corrected or uncorrected)
▪ Commotio cordis
○Primary arrhythmias:
▪ Long QT syndromes
▪ Short QT syndrome
▪ Brugada syndrome
▪ Early repolarization syndromes
▪ Catecholaminergic polymorphic ventricular tachycardia
▪ Idiopathic ventricular fibrillation
▪ Wolff-Parkinson-White syndrome (WPW)
○Non-cardiac causes include:
▪ Sudden death during extreme physical activity
▪ Drug overdose
▪ Toxic/metabolic imbalances (e.g., hyper- or hypokalemia, thyroid storm, adrenergic storm, acidosis)
▪ Acute intracranial hemorrhage
▪ Massive pulmonary embolus
▪ Asthma (or other pulmonary condition)
▪ Aortic dissection
Epidemiology and Clinical Features
○SCD affects 200,000–300,000 per year in the United States (≈0.1% population incidence/year).
▪ It is the initial clinical presentation in up to 20% of patients with CAD.
▪ SCD accounts for up to 50% of CAD deaths.
○The highest proportion of SCD events occurs in the highest-risk subgroups.
▪ Thirty percent of all SCD events occur in the highest-risk subgroup; however, the absolute number of deaths is relatively small owing to the subgroup being very focused.
• This limits the overall population impact of intervention.
▪ Fifty percent of all SCD events occur among subgroups of patients thought to be at relatively low risk for SCD.
• Given the high absolute number of events in this population, the population impact of intervention is potentially great, if these patients could be identified.
Prognosis
○Survival falls rapidly after the initial minutes from the onset of cardiac arrest.
○The likelihood of survival to discharge is 23% for witnessed cardiac arrest, vs. 4% for unwitnessed arrest.
○Recurrence is highest in the first 6–18 months post index event.
IDENTIFYING PATIENTS AT RISK OF SUDDEN CARDIAC DEATH (SCD)
General Risk Stratification
Table 14.1 Factors Affecting Risk of SCD
| Risk of SCD | |
LVSD/HF or previous MI | 5% |
Any two of LVSD/HF, previous MI, or complex ectopy* | 10% |
LVSD/HF + previous MI + complex ectopy | 15% |
Survivor of SCD, or syncopal VT | 20%–40% |
HF: heart failure; LVSD: LV systolic dysfunction (LV ejection fraction [LVEF] <30%–40%); MI: myocardial infarction; SAECG: signal-averaged electrocardiogram (ECG).
*Complex ectopy = >10 premature ventricular contraction (PVC)/h, couplets, triplets, non-sustained ventricular tachycardia (NSVT)
○Predictors of recurrent cardiac arrest in the “survivor” of SCD include:
▪ High brain natriuretic peptide (BNP)
▪ Extensive (multivessel) CAD
▪ Prior MI (within 6 months)
▪ Chronic heart failure (CHF)/LV dysfunction
▪ Ventricular electrical instability (complex ventricular ectopy)
▪ Abnormalities on signal-averaged ECG (SAECG)
Investigations
Table 14.2 Investigations to Determine Risk of SCD
| Parameter | Marker of Risk | Target Group |
Family history | SCD, syncope, or known high-risk cardiomyopathies | All patients |
ECG | NSVT, MI, LQTS/SQTS, Brugada pattern, pre-excitation, possible early repolarization | CAD, LQT, Brugada, WPW HCM |
TWA | Positive TWA | CAD |
SAECG | Positive late potentials | ARVC |
EPS | Short anterograde AP ERP Inducible VT | WPW Cardiomyopathy Bundle branch reentry Tetralogy of Fallot |
Echocardiogram | Low EF Asymmetric LVH | DCM HCM |
Genetic testing | Disease causing mutation Several emerging adverse genetic polymorphisms | LQTS, Brugada HCM ARVC |
ARVC: arrhythmogenic RV cardiomyopathy; DCM: Dilated cardiomyopathy; EPS: electrophysiology study; HCM: hypertrophic cardiomyopathy; LQT: long QT; LQTS: long QT syndrome; LVH: left ventricle hypertrophy; TWA: T-wave alternans.
Signal-Averaged ECG (SAECG)
○SAECG improves the signal-to-noise ratio of a surface ECG, thus facilitating the identification of low-amplitude signals at the end of the QRS.
▪ The late potentials indicate regions of abnormal myocardium, which serve as the substrate for reentrant tachyarrhythmia.
○Abnormal findings include:
▪ Filtered QRS duration (fQRS) ≥114 ms
▪ Root mean-square voltage of terminal 40 ms (RMS40) <20 mcV
▪ Duration of low amplitude signals (<40 μV) in the terminal QRS ≥38 ms
○Interpretation
▪ Presence of abnormal SAECG increases the risk of arrhythmic events 6- to 8-fold post myocardial infarction (MI).
• May signal the need for further risk stratification (i.e., EPS).
▪ High negative predictive value (NPV; 89%–99%).
• Normal SAECG is associated with a <5% change of inducible VT at EPS.
T-Wave Alternans (TWA)
○TWA is a beat-to-beat fluctuation in the amplitude or morphology of the T wave.
○TWA is typically measured on ECG with exercise.
○Its presence identifies high-risk patients (post MI and those with ischemic or non-ischemic dilated cardiomyopathy [NIDCM]).
○Its absence offers good discriminative function (i.e., high NPV).
Heart Rate Variability (HRV)
○HRV is a beat-to-beat variation in cardiac cycle length (CL) due to the autonomic influence on the SN.
▪ It reflects a continuous assessment of the basal sympathovagal influence.
○Derived from 24-hour Holter monitoring.
○HRV independently predicts the risk of SCD and total mortality post MI (with/without LV dysfunction).
Heart Rate Turbulence (HRT)
○HRT is short-term oscillation of cardiac CLs after spontaneous PVCs.
▪ Normally, there is a brief, baroreflex-mediated HR acceleration followed by a gradual deceleration.
▪ In high-risk patients, the typical HRT response is blunted or missing, reflecting reduced baroreflex sensitivity.
○HRT is derived from 24-hour Holter monitoring.
▪ RR intervals surrounding spontaneous PVCs (that fulfill criteria with respect to prematurity and compensatory pause) are averaged to create a “local tachogram.”
○HRT independently predicts the risk of SCD and total mortality post MI (with/without LV dysfunction).
Baroreflex Sensitivity
○Baroreflex sensitivity offers a quantitative assessment of the ability of the autonomic system to respond to acute stimulation.
▪ Most commonly it is performed by analyzing bradycardic response to intravenous phenylephrine bolus.
▪ HR slowing in response to increased blood pressure (BP) indicates the baroreflex tone; a reduced response indicates increased risk.
○Baroreflex sensitivity independently predicts risk of SCD and is additive to HRV and TWA.
Cardiac Meta-Iodobenzylguanidine (MIBG) Scintigraphy or Positron Emission Tomography (PET)
○MIBG indicates sympathetic innervation; PET shows myocardial metabolism.
○Both are possibly better than SAECG, HRV, and QT dispersion at predicting SCD in patients with chronic HF.
Cardiac Magnetic Resonance Imaging (MRI)
○MRI allows for scar quantification and characterization (dense vs. heterogeneous).
○MRI is also useful to identify patients at high risk for ventricular arrhythmias (dilated cardiomyopathy [DCM], HCM, ARVC, post-MI).
Electrophysiology Testing (EPS)
○In general, the positive predictive value (PPV) is about 10% with a NPV of about 95%; however, the overall utility depends on the underlying pathology.
▪ EPS is most useful for ischemic heart disease as well as VT induction in the context of ablation.
▪ EPS for dilated cardiomyopathy or inherited arrhythmia syndromes suffer from the following issues:
• Low inducibility
• Low reproducibility of EPS
• Limited PPV of induced VT
▪ EPS for syncope due to suspected bradyarrhythmia suffers from the following issues:
• Limited sensitivity with episodic bradycardia and syncope
• Common false positive (~25%) and false negative tests
Specific Conditions
CAD
○Epidemiology
▪ CAD is present in 60%–75% of SCD deaths.
• A high proportion of those experiencing SCD have multivessel disease.
• Only 30%–40% of these will have acute infarction.
• It is estimated that 20% of first MI present as SCD.
○Risk factors for SCD:
▪ Risk factor with good sensitivity but poor specificity:
• Reduced LVEF (<40%; particularly if the LVEF is <30%)
▪ Risk factors with good specificity but poor sensitivity include:
• Previous cardiac arrest or a history of aborted SCD
▫ Transmural MI (STEMI): VT/VF <48 h post event does not imply a worse prognosis.
▫ Non-transmural MI (NSTEMI): VT/VF <48 h post event confers increased long-term risk.
• Syncope
• Non-sustained VT (spontaneous)
• Inducible VT at EPS
▫ If LVEF <40%, there is a 35%–45% yearly risk of SCD with an inducible VT at EPS.
▫ If no inducible VT is present at EPS the annual risk of SCD is <5%.
• Other
▫ Late potentials on SAECG
▫ Decreased HRV, microvolt TWA, HRT
▫ Increased QRS duration, QT dispersion, or TWA
Non-Ischemic Dilated Cardiomyopathy (NIDCM)
○Epidemiology
▪ 5-year mortality of ~20%
▪ 30% of all deaths are sudden: VT/VF > bradyarrhythmia
○Risk factors for SCD:
▪ Previous cardiac arrest or a history of aborted SCD
▪ History of syncope
▪ EF <35%
▪ Non-sustained VT
▪ Induction of monomorphic VT at EPS (absence of VT does not confer lower risk)
SUDDENT CARDIAC DEATH (SCD) IN ATHLETES
Incidence
○Annually, 1–3 SCD per 100,000 (RR of 2 to 3 vs. non-athletic peers)
Causes
○HCM (30%–40%)
○Congenital coronary artery anomalies (15%–20%)
○ARVC (5%)
○Ion channel disorders (<5%)
○Autopsy negative (<5%)
○Other causes include:
▪ Myocarditis
▪ Trauma: Commotio cordis or trauma involving structural cardiac injury
▪ Aortic: Ruptured aortic aneurysm, aortic valve stenosis
▪ Atherosclerotic CAD
▪ Asthma (or other pulmonary condition), heat stroke, drug abuse (i.e., cocaine)
Screening
○Annual clinical history (personal and family) and physical examination
○ECG
▪ Common abnormalities in athletes (95%)
• Sinus bradycardia, first-degree AV block
• Notched QRS in V1 (incomplete RBBB), early repolarization, Isolated voltage criteria for left ventricular hypertrophy (LVH)
▪ Uncommon abnormalities in athletes (<5%)
• Chamber enlargement or hypertrophy: Left atrium, right ventricle
• Bundle branch or fascicular block
• Pathologic Q waves, ST segment depression, T-wave inversion
• Brugada-like early repolarization
• Long or short QT interval
• Ventricular arrhythmias
○If the history or ECG is positive, then proceed to further investigations:
▪ ECG, stress test, 24-hour Holter monitor, cardiac MRI, angiogram, EPS
Exercise Restriction
○HCM
▪ Restrict the patient to low-intensity/recreational sports (particularly with obstructive variant).
○Gene carriers without a phenotype (HCM, ARVC, DCM, channelopathies)
▪ Restrict to low-intensity/recreational sports (European Society of Cardiology); no restriction (Bethesda 36).
○Ion channelopathies (QTc >440 ms in men and >460 ms in women):
▪ Restrict to low-intensity/recreational sports.
▪ A recent trend is to favor less restriction, especially if adequately β-blocked.
○Brugada syndrome, catecholamine-induced polymorphic VT
▪ Restrict to low-dynamic/low-static sport.
▪ Recent trend to less restriction
○Marfan syndrome
▪ Restrict to low-intensity/recreational sports unless the aortic root <40 mm.
▪ If < moderate-severe MR and no family history of aortic dissection or SCD, restrict to moderate-intensity competitive sports.
○WPW syndrome
▪ No restriction if asymptomatic (use care in dangerous environments)
▪ Post-ablation may resume competitive sports after 1–3 months
○PVCs or NSVT (<10 beats; <150 bpm; suppresses with exercise)
▪ No restriction if asymptomatic or structurally normal heart
▪ If CV disease, only allowed to participate in low-intensity/non-competitive sports
○ICDs
▪ Restrict to low-intensity/recreational sports without risk of device trauma.
CHANNELOPATHIES
Channelopathies are rare, heritable syndromes.
Long QT Syndrome (LQTS)
General Information
○The incidence of LQTS is 1 in 2500.
○LQTS accounts for 3000–4000 annual sudden deaths in childhood in the United States.
○Mutations are more frequent than the clinical phenotype:
▪ The average QTc penetrance is 25%–60%.
▪ Only 35%-40% of gene carriers are identified by clinical diagnostic criteria.
▪ Exercise testing improves detection and genetic prediction.
Classification
Table 14.3 Classification of LQTS Types
| LQT1 | LQT2 | LQT3 | LQT4 | LQT5 | LQT6 | |
Epidemiology | 40%–55% | 35%–45% | 8%–10% | 3% | 2% | |
Gene/protein | KvLQ1 or KCNQ1 | KCNH2 or HERG | SCN5A | Ankyrin-B | KCNE1 (minK) | KCNE2 (miRP1) |
Channel | Slow delayed rectifier | Rapid delayed rectifier | Sodium channel | Ion channel anchor | Coassembles with KvLQT1 | Coassembles with HERG |
Current | IKs (α) | IKr (α) | INa (α) | INa, IK, INCX | IKs (β) | IKr |
Channel function | ↓ | ↓ | ↑ | ↓ | ↓ | ↓ |
Action potential | Delayed phase 3 | Delayed phase 3 | Prolonged phase 2 | — | Delayed phase 3 | Delayed phase 3 |
Triggers | Exercise Swimming | Emotion Auditory | Rest Sleep | |||
T wave | Broad T Late onset | Bifid T Low amplitude | Asymmetric, late and peaked | |||
Epinephrine or isoproterenol | ↑ QT | ↓ QT | ↓ QT | |||
Mexilitine | — | — | ↓ QT | |||
Events <10y | 40% | 16% | 2% |
○LQT7 – Andersen-Tawil syndrome
▪ KCNJ2 mutation leads to loss of function alters inward rectifier K current through the Kir2.1 channel.
▪ Clinical
• Potassium-sensitive periodic paralysis
• Dysmorphic features: Short stature, hypertelorism, palate defect, broad nasal root
▪ ECG: Pseudo long QT with prominent U wave
▪ Ventricular arrhythmias: Very large PVC burden (up to 50% ectopy), bidirectional VT
▪ Prognosis: Benign
Epidemiology and Clinical Features
○LQTS is mostly asymptomatic.
○Common symptoms include syncope, seizures, and cardiac arrest.
○Associated symptoms may include:
▪ Sensorineural deafness (Jervell & Lange-Nielsen: Autosomal recessive)
▪ Periodic paralysis (Andersen-Tawil: Autosomal dominant heterozygote)
○Family history
▪ Positive for LQT or SCD
12-Lead ECG
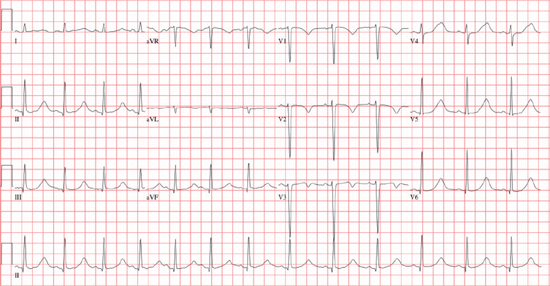
○Measuring the QT interval
▪ Average QT and RR interval over ≥3 QRS complexes in ≥3 ECG leads.
▪ Measure from the onset of the QRS complex to the end of the T wave (the point where the tangential line from the steepest terminal portion of the T wave crosses the isoelectric line).
▪ Corrected QT (QTc)
• Bazett’s formula: QTc = QT/√(RR in seconds)
• Normal: 390–450 ms (men) or 390–460 ms (women)
• Most borderline prolonged intervals are normal when repeated.
Diagnosis: Schwartz Score
○The Schwartz score combines ECG and clinical parameters to estimate the probability of inherited LQTS with high specificity (80%–100%) but low sensitivity (70% for score ≥4; >30% for <4).
▪ ECG parameters include:
• QTc: ≥480 ms: 3 points; 460–470 ms: 2 points; >450–460 ms (males): 1 point
• Torsade de pointes: 2 points
• T-wave alternans: 1 point
• Notched T wave in three leads: 1 point
• Resting HR below second percentile for age (children): 0.5 point
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


