Sudden Cardiac Death and Implantable Cardioverter-Defibrillators
Frank Pelosi Jr.
Fred Morady
Overview
More patients die of sudden cardiac death (SCD) annually than from lung, breast, and colon cancer combined. SCD is usually associated with significant structural heart disease, but it can also be caused by ion channel disorders in patients with otherwise normal hearts. Treatment of cardiac arrest begins with resuscitative measures, followed by steps to prevent its recurrence. Antiarrhythmic drugs to prevent SCD have been eclipsed by the implantable cardioverter-defibrillator (ICD) to treat ventricular arrhythmias that are the most common cause of SCD. This chapter reviews various aspects of SCD, as well as evaluation and management of patients who survive aborted SCD episodes.
Definition
SCD is defined as unexpected death of a primarily cardiac origin, usually within 1 hour of symptom onset (1). Many associated conditions can be present that dramatically increase the risk of SCD, although, in many patients, no such condition is found.
The definition of SCD has important implications for analyzing the impact of sudden death on society. Previous definitions defined SCD as death within 24 hours of the onset of symptoms or out-of-hospital death or death on arrival to an emergency department. Such definitions make determination of the actual cause of death difficult without an autopsy. Even then, death certificate data have been shown to overestimate the incidence of SCD by 24% (2).
Epidemiology
Approximately 335,000 people die of SCD per year, accounting for almost half of those who died of coronary heart disease (3). Half of these patients have no previously diagnosed heart disease. The death rate from SCD increases with age, although younger patients have a higher proportion of SCD relative to death from other cardiac causes (4). In younger age groups, men younger than 55 years of age have a death rate from SCD of 140 per 100,000 population, versus 45 per 100,000 in women in the same age group. In older age groups, this gender difference decreases until women’s death rate overtakes that of men at about age 70, to 4,100 per 100,000 population (4). This is probably because women develop coronary artery disease at a later age than men and are less likely to survive when cardiac arrest occurs at an older age.
Among ethnic groups in the United States, African Americans have a higher death rate from SCD than other ethnic groups (4). Death rates in Asian Americans and Hispanic Americans may be underestimated because of inaccuracies in U.S. Census data. The reasons for this disparity remain unclear.
As is the case with other cardiac disease, the death rate from SCD is decreasing. Data from the Framingham Study population indicate that the death rate from SCD has decreased by 49% over the past 50 years, somewhat less than the 64% decrease for deaths from other cardiac disease (5). These decreases were found in both patients with and without antecedent cardiac disease at the time of death. Primary prevention measures, such as lipid-lowering therapy, blood pressure control, and increased aspirin and defibrillator use, may contribute to the
reduction. The increased availability of rapid resuscitation also may be responsible for improvements in survival.
reduction. The increased availability of rapid resuscitation also may be responsible for improvements in survival.
Mechanisms of Sudden Cardiac Death
In the few studies in which ambulatory monitoring captured cardiac arrhythmias responsible for sudden cardiac death, ventricular tachycardia and fibrillation caused most SCDs, followed by profound bradycardia (6,7,8). Sinus node dysfunction and supraventricular tachyarrhythmias are rare causes of SCD, but these conditions may be fatal in the presence of severe obstructive lesions such as aortic or mitral stenosis or left ventricular outflow obstruction.
Ischemia- and Reperfusion-Induced Ventricular Arrhythmias
Myocardial ischemia results in depolarization of the resting membrane potential because of increases in extracellular potassium concentration from acidosis and low adenosine triphosphate (ATP) concentrations as well as accumulation of intracellular metabolites such as lysophosphoglycerides (9,10). In addition, increased inactivation of myocardial sodium channels decreases the action potential amplitude and upstroke velocity. The action potential response to ischemia varies across different myocardial cell types, with subepicardial cells having more reduced upstroke velocities than subendocardial cells (11). Action potential duration is affected by the changes in membrane potential and the duration of coronary occlusion, initially lengthening, then shortening with prolonged ischemia (12).
Variations among different cell types and tissue regions result in heterogeneous changes in action potential characteristics and refractory periods when exposed to ischemia. The “border zone” around ischemic myocardium demonstrates shorter refractory periods than normal myocardium, whereas the infracted zone has longer refractory periods (13).
Experimental studies suggest that premature ventricular depolarizations are attributable to a reentrant mechanism during the earliest phases of ischemia (14). Ventricular tachycardia can occur when premature ventricular depolarizations interact with myocardial tissue with differing electrophysiologic properties, with resulting unidirectional conduction delay in the ischemic border zone (15). Fragmentation of these circuits can produce ventricular fibrillation. Even after prolonged cardiac ischemia, surviving Purkinje cells exhibit electrophysiologic properties that can provide the substrate for ventricular arrhythmias up to 72 hours after infarction has occurred, and this mechanism is thought to account for ventricular arrhythmias late after myocardial infarction (16).
Reperfusion of ischemic myocardium can also trigger ventricular arrhythmias. Reperfusion causes an immediate washout of extracellular potassium and metabolites, resulting in heterogeneous changes in action potential properties in previously ischemic and normal myocardial tissue (16).
Autonomic Influences in Ventricular Arrhythmias
Heightened sympathetic tone contributes to the arrhythmic substrate during ischemia. Increased sympathetic stimulation increases heart rate and ischemic burden and directly affects sodium potassium and calcium currents. During ischemia, increases in action potential upstroke velocity, duration, and amplitude are caused by accumulation of norepinephrine and increases in potassium conductance and sodium/potassium ATPase activity (17). Reductions in ischemia-induced action potential alterations have been observed after exposure to propranolol and experimental denervation (17,18).
Clinical trials suggest that an increase in sympathetic activity predisposes patients to ventricular arrhythmias. Heart rate variability and baroreflex sensitivity were used to assess autonomic tone in the Autonomic Tone and Reflexes After Myocardial Infarction (ATRAMI) trial. In this trial, 1,284 patients who had a history of myocardial infarction were followed for a mean of 21 months. Two-year mortality for those with depressed heart rate variability and baroreflex sensitivity was 17% versus 2% in those with preserved heart variability and diminished left ventricular ejection fraction (19). Therapies such as β-blockers and angiotensin-converting enzyme antagonists have been shown to modulate sympathetic tone and to increase heart rate variability. Several clinical trials have demonstrated that alterations of heart rate variability with these agents have a beneficial effect on mortality.
Causes of Sudden Cardiac Death
The causes of SCD are shown in Table 67.1.
TABLE 67.1 Clinical Conditions Associated with Sudden Cardiac Death | |
|---|---|
|
Coronary Artery Disease
Coronary artery disease is the most common condition associated with SCD, present in 30% to 70% of cardiac arrest survivors. SCD associated with coronary artery disease generally occurs during one of two clinical scenarios.
Acute occlusion of a major coronary artery results in the electrophysiologic changes described previously (see the section on pathophysiology of SCD). Autopsy studies have shown that approximately 20% of patients have evidence of acute myocardial infarction, almost 90% of whom had active coronary occlusions or plaque rupture at the time of death (20). Histologic and gross examinations show that myocardial infarctions are divided roughly equally between subendocardial and transmural infarctions. Because of the rapid onset of death, evidence of myocardial scar may not be evident at autopsy. Healed myocardial infarction alone is seen in approximately 40% of cases. With this presentation, only 45% of patients have active coronary lesions at the time of death. Many of these patients have no evidence of ischemia before their demise, but they have scars that provide the electrophysiologic substrate for ventricular arrhythmias leading to sudden death (21).
Nonischemic Dilated Cardiomyopathy
Nonischemic or dilated cardiomyopathy is defined as chronically diminished left ventricular systolic function in the absence of significant coronary artery disease or myocardial infarction. Nonischemic cardiomyopathy accounts for 18% of patients found to have a left ventricular ejection fraction of less than 0.40 in the United States (22). Unlike ischemic cardiomyopathy, nonischemic cardiomyopathy is characterized by patchy, diffuse fibrosis and subendocardial scarring.
Hypertrophic cardiomyopathy is characterized by thickening of the left ventricle, most commonly the interventricular septum, and preserved systolic function, although this function may deteriorate as the cardiomyopathy progresses. Several reports have established that hypertrophic cardiomyopathy is associated with an increased risk of SCD, particularly when hypertrophy is severe (23,24). Implantable defibrillators have been shown to be highly effective in terminating ventricular arrhythmias associated with this condition (25).
Arrhythmogenic right ventricular dysplasia is characterized by fatty infiltration and fibrotic replacement of the myocardium of the right ventricle, although the interventricular septum and left ventricle can also be involved (26,27). Features include right ventricular dilation and dysfunction, abnormal electrocardiographic findings in the anterior precordial leads, and a variety of ventricular arrhythmias (28). This syndrome is associated with an increased risk of SCD, and up to 75% of patients who receive an ICD have appropriate therapies for ventricular arrhythmias (29).
Other infiltrative diseases are associated with SCD. Cardiac amyloidosis is commonly associated with SCD as amyloid proteins infiltrate myocardial tissue and coronary microvasculature (30,31,32). Ventricular arrhythmias and abnormalities of atrioventricular conduction have been observed at electrophysiologic testing (33). Ventricular tachycardia in patients with cardiac sarcoidosis is believed to involve active or healing granulomatous lesions (34). In small study populations, several reports describe successful termination of ventricular tachycardia with an implantable defibrillator, a finding suggesting that defibrillators are an effective therapy for patients with cardiac sarcoidosis and ventricular arrhythmias (35,36). Myocarditis can be caused by infectious or autoimmune conditions and may result in otherwise unexplained sudden death with minimal antecedent symptoms (37,38,39,40,41). Histologic features include subepicardial infiltrations of lymphocytes, plasma cells, multinucleated giant cells, or monocytes with central myocardial necrosis (39,42).
Genetic Disorders
Long QT syndrome is a genetic disorder affecting 1 in 5,000 persons and is typified by a prolonged QT interval and life-threatening ventricular arrhythmias. Currently, hundreds of mutations have been described on 8 genes regulating cardiac ion channels. Most mutations (LQT1, 2, 5, and 6) have been found on genes controlling potassium ion channel subunits, α and β. Less common mutations (LQT3) are found in sodium channel subunits and rarely in calcium channel subunits. However, a specific mutation cannot be identified in 30% to 35% of patients with long QT syndrome. Depending on the genotype, 10% to 16% of patients with long QT syndrome will die or experience cardiac arrest before the age of 40 years if they are not treated (43). β-Blockers attenuate sympathetic tone that can trigger ventricular arrhythmias. Registry data suggest that β-blockers reduce cardiac events in mildly symptomatic patients and affected relatives (44). Patients with severe symptoms such as syncope or cardiac arrest also benefit, but they continue to have events. An ICD usually is appropriate for these patients. Patients can manifest electrocardiographic and clinical signs of long QT syndrome when they are exposed to a wide variety of medications. These patients have a subclinical form of long QT syndrome that is unmasked by a drug that affects cardiac repolarization.
Short QT syndrome is less common and less well studied than its long QT counterpart. Families have been identified with recurring aborted SCD and persistently short QT intervals (<300 milliseconds). Mutations on three different genes that control the potassium current (IKr) have been identified (45). Interestingly, this mutation results in a gain of function of the ion channel. In contrast, in the long QT syndrome, mutations of IKr cause a loss of function. Quinidine, a drug that prolongs the QT interval, has been shown to reduce the inducibility of ventricular tachycardia in affected patients (46).
The Brugada syndrome was first described in 1992 in a group of young patients with recurrent cardiac arrest caused by polymorphic ventricular tachycardia (47). These patients typically display coved ST-segment elevation in V1 and V2 (Fig. 67.1). These abnormalities are caused by altered sodium channel physiology in phase 2 of the action potential. Genetic studies have linked this syndrome with mutations in the SCN5A gene; however, up to 75% of affected patients have no such mutation. Risk stratification is based on the presenting symptoms, the baseline electrocardiographic abnormalities, and the inducibility of ventricular tachycardia at electrophysiologic study. ICDs are recommended for high-risk patients.
Idiopathic ventricular fibrillation applies to those patients in whom ventricular fibrillation occurs with no clear cause. Several genetic mutations have been implicated in this disorder, but some overlap with other established syndromes, such as the Brugada or short QT syndromes. During electrophysiologic studies, closely coupled premature ventricular contractions seem to trigger ventricular fibrillation. Successful radiofrequency ablation of the premature depolarizations that trigger ventricle fibrillation may prevent recurrences, but an ICD generally is appropriate for reliable prevention of SCD (48,49,50).
Electrolyte Abnormalities
Several electrolyte abnormalities can contribute to cardiac arrest or can affect resuscitative efforts. Hyperkalemia can occur
from exogenous causes, such as medications, or as a complication of renal failure or hypoaldosteronism. Potassium concentrations greater than 6 mEq/L can cause early electrocardiographic changes such as tall, peaked T waves and first-degree atrioventricular block. Further elevations in potassium concentrations (>7 mEq/L) can lead to a sine wave pattern on the electrocardiogram, idioventricular rhythms, and asystole. Hypokalemia (<3.5 mEq/L) can produce T-wave flattening or U-waves on electrograms and ventricular arrhythmias, especially in the presence of other medications, such as digoxin.
from exogenous causes, such as medications, or as a complication of renal failure or hypoaldosteronism. Potassium concentrations greater than 6 mEq/L can cause early electrocardiographic changes such as tall, peaked T waves and first-degree atrioventricular block. Further elevations in potassium concentrations (>7 mEq/L) can lead to a sine wave pattern on the electrocardiogram, idioventricular rhythms, and asystole. Hypokalemia (<3.5 mEq/L) can produce T-wave flattening or U-waves on electrograms and ventricular arrhythmias, especially in the presence of other medications, such as digoxin.
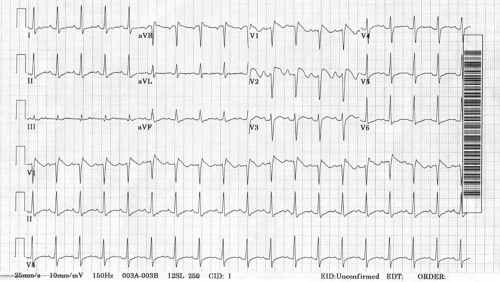 FIGURE 67.1. ST elevation in V1 and V2 typical of the Brugada syndrome. |
Hypomagnesemia (<1.3 mEq/L) is associated with frequent premature ventricular depolarizations, QT prolongation, and polymorphic ventricular tachycardia, known as torsades de pointes (51




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
