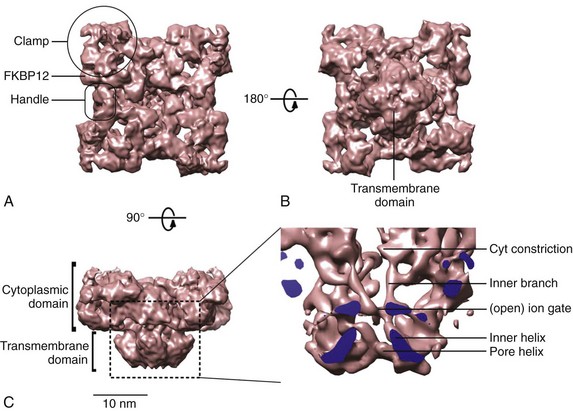
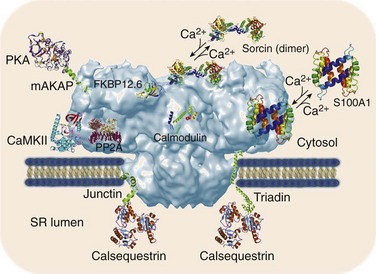
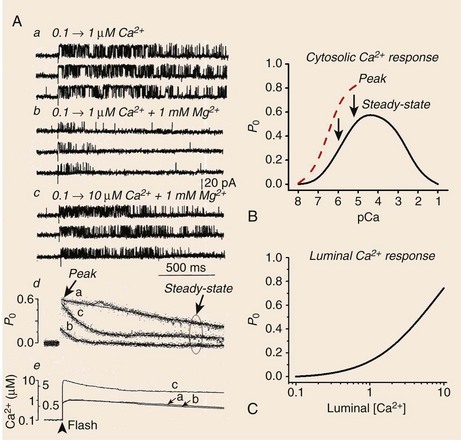
6 The sarcoplasmic reticulum (SR) is a membrane-delimited intracellular organelle present in striated muscle of almost all species.1 Its name literally means “network of structures located in the cytoplasm of a muscle fiber,” referencing the interconnected network of tubules and vesicles that spans the sarcomere and wraps up the contractile myofilaments. The SR is not continuous with the external membrane, but recent data indicate that it is continuous with the nuclear envelope.2 In cardiac and skeletal muscle, the main function of the SR is to provide the majority of Ca2+ ions that are needed to activate the contractile proteins of the myofilaments, and to resequester Ca2+ from the myoplasm to allow for relaxation. The Ca2+ concentration within the SR ([Ca2+]SR ) is approximately 1 mM, with a larger pool of Ca2+ bound to calsequestrin and other Ca2+-binding proteins.1 Therefore, when myofilaments are relaxed and bathed in cytoplasmic milieu containing approximately 0.1 µM Ca2+, there is an approximately 10,000-fold [Ca2+] gradient across the SR membrane. The compartmentalization of muscle fibers into small (~2 µm) structural-functional units (sarcomeres) ensures that Ca2+ diffusion from release sites is not a limiting step in muscle contraction. Similarly, the enveloping of myofilaments by the SR ensures rapid reuptake of Ca2+, leaving rate-limit steps of relaxation to contractile proteins. It is useful to compare the structural arrangement of the SR in cardiac and skeletal muscle to facilitate discussion of the mechanisms that trigger Ca2+ release in these two muscles. The series of events by which depolarization of the sarcolemma generates a mechanical contraction is termed excitation-contraction coupling (E-C coupling). Although common subcellular structures participate in E-C coupling, different processes link membrane depolarization to Ca2+ release in cardiac and skeletal muscle. Some of these differences stem in part from the ultrastructural arrangement of the muscle fiber’s organelles. It is well accepted that E-C coupling in skeletal muscle is greatly dependent on Ca2+ release from the SR, with little or no Ca2+ entry across the sarcolemma during a single twitch. Accordingly, the SR of skeletal muscle is robust and contains large saccular enlargements, whereas T-tubules, which are invaginations of the external membrane that carry the depolarizing stimulus to the interior of the cell, are narrow (~20 to 40 nm) and elongated. By contrast, Ca2+ entry across the sarcolemma is an obligatory step for E-C coupling in cardiac muscle.3 T-tubules, which not only carry the electrical stimulus to the interior of the cell but must serve also as Ca2+ reservoirs, are therefore larger in diameter (~200 nm) in cardiac muscle. The SR appears thinner and less organized than in skeletal muscle, but exhibits multiple points of contact with the sarcolemma and T-tubules.4 Depending on the animal species and the type of cardiac cell, the SR volume can be as high as 12% of cell volume (mouse atrium), to as little as 0.8% (finch ventricle).1 These sharp differences likely reflect the relative importance of Ca2+ entry versus SR Ca2+ release for contraction among different cells: whereas mouse cardiomyocytes rely heavily on Ca2+ release from the SR, finch (and all avian) ventricular cells lack T-tubules altogether and thus rely almost entirely on Ca2+ fluxes across the sarcolemma. In both cardiac and skeletal muscle, the L-type Ca2+ channels or dihydropyridine receptors (DHPRs) are the voltage sensors of sarcolemma and T-tubules that initiate E-C coupling, and the SR Ca2+ release channels that provide the majority of Ca2+ for contraction are also known as ryanodine receptors (RyRs).4 The manner in which DHPRs trigger RyRs to open is particular to each muscle. Because skeletal muscle fibers can twitch in the absence of external Ca2+, and Ca2+ release follows sarcolemmal depolarization with virtually no delay, a physical coupling between DHPRs and RyRs is thought to initiate E-C coupling in skeletal muscle.5 Indeed, identification of the structural domains of DHPRs and RyRs involved in their mechanical interaction continues in earnest,6 although the presence of proteins “sandwiched” between these channels has not been completely ruled out. By contrast, RyRs of cardiac muscle are closely apposed but do not appear mechanically joined to DHPRs4; instead, they are activated by a small influx of external Ca2+ (the inward Ca2+ current provided by DHPRs, or ICa), releasing greater amount of Ca2+ from the SR.7 This process, Ca2+-induced Ca2+ release (CICR),8 is a signature event of RyRs but is not exclusive to cardiac muscle. In fact, CICR was first described in skeletal muscle,9 where it apparently serves to activate RyRs that are not coupled to DHPRs.10 Again, the structural arrangement of DHPRs and RyRs is congruent with the type of E-C coupling they exert in skeletal and cardiac muscle. In the former, a great proportion of DHPRs appears organized as tetrads that rest on top of a single RyR channel in the junctional SR, the specialized region of the SR that is proximal to T-tubules.4 Because RyRs are homotetrameric protein complexes (discussed later), each DHPR in a tetrad is presumed to interact with one RyR monomer. RyRs, on the other hand, are clustered in a paracrystalline lattice where they touch each other at the corners.11 In this orderly array, every other RyR interacts with a tetrad of DHPRs and is directly activated by the physical coupling mechanism; the DHPR-free RyR is presumably activated by CICR and cooperative interaction between RyRs.4,10 Multiple (i.e., 10 to 20) such DHPR-RyR interactions occur in a couplon, the structural domain of the junctional SR where E-C coupling actually takes place.12 In cardiac muscle, DHPRs are irregularly and sparsely distributed (not forming tetrads) in a couplon, but RyRs keep their lattice array, thus changing the stoichiometry and the mode of interaction of these two channels. A typical couplon in cardiac myocytes can contain approximately 100 RyRs and only approximately 20 DHPRs.1 Thus, in principle, one DHPR injects Ca2+ into a couplon to activate approximately 5 RyRs, but current estimates indicate that E-C coupling is normally effected with a high safety factor—that is, only a portion of the existing DHPRs is needed to fully activate RyRs13 (discussed later ). In addition to the junctional SR, DHPRs and RyRs interact at the peripheral couplings, the points of contact between SR and sarcolemmal surface.4 Despite the fact that T-tubules contain the highest density of DHPRs, couplons of the peripheral couplings apparently have DHPR/RyR stoichiometry that is similar to those of junctional SR couplons. Thus, RyRs of peripheral couplings are expected to release Ca2+ by a model similar to that described earlier for RyRs of junctional SR, although this has not been tested rigorously. A different mechanism, however, must operate to activate RyRs in the corbular SR, a basketlike form of SR that is connected to the SR network at one point only, and completely lacking T-tubular or sarcolemmal contacts. Activation of corbular RyR channels probably requires a combination of Ca2+ diffusion from release sites and a propagating wave of CICR. Corbular SR can contain up to approximately 35% of the total RyRs and is particularly prominent in atrial cells and Purkinje fibers.14 Advancements in confocal imaging techniques have started to shed new light on SR ion channels in their cellular environment, but at present the majority of their functional and biochemical properties have been defined using isolated SR vesicles. Homogenization of cardiac and skeletal muscle produces fragmented SR that can be partially segregated by sucrose density centrifugation into RyR-enriched “heavy” SR (corresponding to junctional SR and peripheral couplings), and Ca2+ pump–enriched “light” SR (corresponding to longitudinal SR). Because RyRs are confined to regions of the cell that are largely inaccessible to patch electrodes, reconstitution of heavy SR into artificial lipid bilayers remains the most versatile and effective method to characterize the biophysical and pharmacologic properties of RyRs at the single channel level.15,16 A vast body of knowledge has already been gained with this technique,16,17 which remains the method of choice in situations that require precise control of modulators on both faces of the channel (e.g., in cytosolic or luminal Ca2+ titrations), and in measurements of unitary channel conductance under quasi-physiologic conditions. In addition, defining the potency and mechanism (open- or closed-channel block), which is indispensable for the rational design of drugs for various RyR-linked diseases, would be extremely difficult without single-channel recordings in lipid bilayers. Nevertheless, it is important to recognize the limitations of this technique, most of which stem from the extremely difficult recreation of the conditions under which RyRs operate in situ. In their intracellular environment, RyRs are activated by fast and transient Ca2+ stimuli (as opposed to stationary Ca2+ levels as is routinely used in lipid bilayer experiments), which modify RyR activity importantly (discussed further later). Furthermore, RyRs seldom work in isolation, as in single-channel experiments, but are clustered in arrays that exhibit cooperativity and synergism.11,12 Lastly, various accessory proteins of the RyR control its response to cytosolic and luminal Ca2+, and these proteins might not be present in reconstituted channels. The optical detection of Ca2+ fluxes has provided new insight into the working principles of the supramolecular Ca2+ signaling system of many cells. The discrete, transient, and presumably elemental Ca2+ signaling events of cardiac myocytes, also known as Ca2+ sparks, are indisputable signs of RyR gating in situ.18 The introduction of fluorescein- and rhodamine-based Ca2+ indicators of high dynamic range and the arrival of low-cost versatile confocal microscopes greatly facilitated the discovery of Ca2+ sparks, first detected in ventricular myocytes19 and later in smooth20 and skeletal21 muscle cells. The low myoplasmic [Ca2+] of resting cells keeps the open probability (Po) of RyRs extremely low; still, RyR channels open with a finite rate that depends on several factors (most notably, [Ca2+] on the cytoplasmic and luminal side of the RyR), giving rise to spontaneous Ca2+ sparks. An estimate of spontaneous spark rates in ventricular myocytes was initially 100/cell/sec,18 which suggested an opening rate for RyRs of 10−4 s−1, assuming that a typical ventricular myocyte contains approximately 1 million RyRs.19 However, spark rates vary widely among investigators apparently using the same conditions and can even be influenced by experimental artifacts such as cell damage during isolation, making this estimate less reliable. Initially, Ca2+ sparks were thought to originate from the opening of a single RyR,18 but the unitary RyR channel conductance in near physiologic conditions (~0.5 pA)22 appears to be too low to deliver the fluorescence signal mass of a typical Ca2+ spark.23 Additional studies detected smaller RyR-originated Ca2+ signals (e.g., “Ca2+ quarks,”24 “Ca2+ embers,”25 “Ca2+ syntilla,”26), somewhat demoting the elemental adjective bestowed on Ca2+ sparks as indivisible events of E-C coupling in cardiac cells. Because Ca2+ sparks typically give rise to a twofold increase in fluorescence intensity in an area of approximately 2 µm, it is likely that they result from the coordinated opening of a portion of (or all) RyRs that are clustered in a couplon (~100). Regardless of the number of RyRs that intervene to form a Ca2+ spark, these Ca2+ signaling events have brought fresh insights into the mechanisms that modulate the activity of RyRs in their intracellular environment. Measurements of RyR density and activity can be readily obtained by performing [3H]ryanodine binding assays in purified SR vesicles or even in whole-tissue homogenates. This is possible because of the high affinity and specificity of [3H]ryanodine for its receptor, which yield an excellent signal-to-noise ratio, and because the alkaloid binds to open RyR channels only.27 The binding of [3H]ryanodine is enhanced by activators of Ca2+ release (Ca2+, adenosine triphosphate [ATP], caffeine) and decreased by inhibitors of Ca2+ release (Mg2+, H+, calmodulin), suggesting that the alkaloid binds to a conformationally sensitive domain of the RyR protein.28 Therefore, [3H]ryanodine can be used as a probe of the functional state of the RyR channel. This approach has contributed to the purification of the RyR itself and to the identification of novel ligands, endogenous modulators, and accessory proteins of RyRs17,27,29; however, ryanodine also displays some disadvantages as ligand of RyRs. Mainly, ryanodine has slow association and dissociation rates, which require long incubation times to reach equilibrium (>15 h at room temperature) and predisposes the RyR to protein degradation. Furthermore, its sluggishness to bind to its receptor renders ryanodine incapable of tracking dynamic changes in RyR activity, which are usually transient and swift (discussed later). Despite these drawbacks, [3H]ryanodine binding is the assay of choice to measure the averaged activity of literally billions of RyRs at the same time and under multiple conditions. Ca2+ movements across the SR can be measured in situ by electron probe analysis of ultrathin cryosections of muscle rapidly frozen at different times in the contraction and relaxation cycle.30 These pioneering experiments have provided critical data on the spatial and temporal movement of Ca2+ and its countercharge ions (mainly K+ and Mg2+) across the distinct regions of SR during the E-C coupling cycle. Foundational notions, such as the SR undergoing no charge deficit during Ca2+ release (and thus maintaining no transmembrane potential) and the terminal cisternae being the main Ca2+ release site of the SR, were derived largely from electron probe analysis experiments.30 RyRs are the main pathway for Ca2+ release from the SR, but they are not restricted to striated muscle. To gain the functional flexibility necessary to respond to different triggering signals, at least three isoforms of RyR are expressed in mammals.17,29,31 RyR1 is expressed predominantly in fast- and slow-twitch skeletal muscle and in cerebellar Purkinje cells. RyR2 is found in cardiac muscle (presumably the only isoform expressed there), but is also robustly expressed in the brain and in visceral and arterial smooth muscle. RyR3 is the least understood of the RyR isoforms and appears to play its most important role during development, although in mature cells it is found in the diaphragm, epithelial cells, brain, and smooth muscle. Several structural and functional characteristics confer to RyRs, a distinctive earmark. RyRs are homotetramers of large molecular size (~2 million Da); they form Ca2+-gated Ca2+-permeable channels of large conductance,32 and they are distinctively affected by the plant alkaloid ryanodine.27 Elucidating the structure of RyRs has been difficult because of the channel’s massive size; however, some details of RyR structure have been obtained through cryoelectron microscopy,6,33,34 comparative modeling,35 and recently x-ray crystallography of small RyR segments.36,37 In electron micrographs, RyRs are seen as quatrefoil or cloverleaf-shaped structures,33,34,38 or in three-dimensional renderings as mushroom-shaped structures, with a large (27 × 27 × 12 nm) cytoplasmic assembly and a smaller transmembrane “stalk” spanning approximately 6.5 nm from the base of the cytoplasmic region and extending into the SR lumen38 (Figure 6-1). The carboxyl-terminal segment crosses the SR membrane as few as four and as many as 10 times (depending on the model, although the consensus is for six transmembrane domains) and forms the Ca2+-permeable pore, whereas the bulk of the protein (~90%) protrudes into the cytosol to bridge a 15- to 20-nm gap between the SR and T-tubule membranes. The quatrefoil structure results from the symmetric arrangement of four identical subunits of approximately 5000 amino acids each; therefore, a single tetrameric channel encompasses approximately 20,000 amino acids. Figure 6-1 Three-dimensional surface representations of the ryanodine receptor (RyR) channel. Surface representations of the RyR1 at 10 Å resolution as seen from the T-tubule (A), the SR side (B), and from the side (orthogonal to the SR membrane) (C). The inset is the magnified region indicated by the square, at a higher density threshold, and cut through the fourfold axis to better visualize the ion pathway. The cutting plane is indicated in blue. (Courtesy M. Samsó, Virginia Commonwealth University, Richmond, Va.). The RyR2 channel can be viewed as a molecular switchboard that integrates a multitude of cytosolic signals, such as dynamic and steady Ca2+ fluctuations, β-adrenergic stimulation (phosphorylation), oxidation, and metabolic states and that transduce these cytosolic signals to the transmembrane domain to release appropriate amounts of Ca2+. Furthermore, Ca2+ release is critically influenced by luminal (intra-SR) factors such as Ca2+ content and protein interactions, thus conferring RyR2 channels an additional role as integrative switch-valves that offset cytosolic-luminal Ca2+ imbalances. Most of the signal-decoding structures are integral domains of the RyR2 protein. In addition to the huge structural tetrameric assembly being highly complex, RyR2 channels are also capable of protein-protein interactions that allow them to bind, in some cases steadily and in other cases in a time- and Ca2+-dependent manner, to small and independently regulated accessory proteins that add another layer of versatility (and complexity) to regulation of Ca2+ release in vivo (Figure 6-2). The best-known RyR2-interacting proteins are calmodulin, which tonically inhibits Ca2+ release39,40; FKBP12.6, which presumably stabilizes RyR2 closures41,42,43; sorcin, which inhibits Ca2+ release in a Ca2+-dependent manner44–46; and the ternary complex triadin-junctin-calsequestrin, which “senses” luminal Ca2+ content and modulates RyR2 activity by acting either as a direct channel ligand or as an immediate source of releasable Ca2+.47,48 S100A1,49 like calmodulin and sorcin, inhibits RyR2 more conspicuously when [Ca2+] is high; therefore these three proteins might play a role in Ca2+-mediated CICR termination. More recently, RyR2 has been found to hold anchoring sites for PKA, PP2A, cyclic adenosine monophosphate (cAMP)-specific phosphodiesterase (PDE4D3) and CaMKII,50 underscoring the importance of RyR2 regulation by phosphorylation. Thus, the RyR2 protein is not only capable of forming ordered multi-channel arrays,4,11 but is also at the center of a massive macromolecular complex that includes numerous regulatory proteins. Although the intrinsic function of each of these accessory proteins is known to some extent, it is still unclear how this complex of proteins interplays in situ to regulate Ca2+ release. Figure 6-2 Three-dimensional model of the ryanodine receptor (RyR) supramolecular complex. The faded blue structure is the cryo-electron microscopy surface representation of the RyR1 protein at 10 Å resolution as shown in Figure 1, C. The multi-colored proteins are represented by their actual crystal structures. Junctin and triadin were generated by Song et al.136 using homology modeling. Binding sites for FKBP12.6 and calmodulin are based on cryo-EM data.33,34 The other protein interacting sites are idealized. Only some of the most relevant proteins that interact with the RyR2 channel are shown. It is necessary to discuss the effect of Ca2+ on isolated RyRs before considering the intricacies of Ca2+ regulation of RyRs in situ. Under stationary (nonfluctuating) [Ca2+], the activity of single RyR2 channels is a bell-shaped function of cytosolic [Ca2+] (Figure 6-3) because of the presence of Ca2+-activating and inactivating sites.17,29,51 Ca2+ in the range of 0.1 to 10 µM binds to at least one Ca2+-binding domain that activates RyR2s. Higher [Ca2+] (100 µM to 3 mM) then inactivates the channel.51 The affinity and cooperativity of the activating and inactivating Ca2+-binding sites vary greatly under the presence of other relevant modulators (e.g., ATP, Mg2+, H+) and constitute powerful mechanisms by which posttranslational modifications of the channel protein (e.g., oxidation, phosphorylation, nitrosylation) modulate Ca2+ release. Among the RyR isoforms, RyR2 is particularly recalcitrant to Ca2+-dependent inactivation, requiring supraphysiologic [Ca2+] (~10 mM) for complete inactivation.17,29,51 For this reason, the functional role of this process is questionable, although its participation in CICR termination has not been ruled out (discussed later). This overall picture represents the response of RyR2 channels to stationary levels of Ca2+, but is different from that obtained under dynamic (fast and transient) Ca2+ stimuli. In what are now landmark studies delineating CICR, Fabiato8,52 observed that the magnitude of Ca2+ release was dependent on the velocity of the Ca2+ pulse (d[Ca2+]/dt) applied to skinned cardiac fibers. Similarly, single-channel studies have revealed that a fast Ca2+ stimulus elicits a transient increase in the Po of RyR2s. If Ca2+ is applied rapidly but remains sustained for long periods, RyR2 Po will relax, or adapt, to a new steady state that follows the activation curve obtained under stationary [Ca2+]. Thus, Po–[Ca2+] curves of greater magnitude emerge from titration of RyR2 activity with fast, calibrated Ca2+ pulses compared with those obtained under similar but steady applications of Ca2+ (Figure 6-3).53–55 The implication of these findings is that there are components of RyR2 activity that remain undetected in most of the assays presently used, and these aspects of RyR regulation might explain seemingly discrepant results, as in the case of RyR2 phosphorylation (discussed later). Figure 6-3 Modulation of RyR2 channel by Ca2+. (A) Activation of RyR2 by fast changes of the [Ca2+] surrounding the cytosolic side of the channel. The resting [Ca2+] was 0.1 µM in all traces. Calibrated steps increases of [Ca2+] were achieved by laser photolysis of caged Ca2+ (nitrophenyl-ethylene glycol tetraacetic acid [NP-EGTA]), as described.54 The RyR2 openings were elicited by fast increase of [Ca2+] to 1 µM (a and b) or to 10 µM (c) produced by single laser pulses. Mg2+ (1 mM) was present in b and c. Traces in all panels were recorded from the same channel. (d) Ensemble currents were generated by the sum data of multiple sweeps. (e) The amplitude and time course of the change in [Ca2+] in the cytosolic side of the channel. (B) Ca2+–Po curves of RyR2 channels. Activity was measured at the indicated stationary concentrations of Ca2+ (steady-state) and right after a fast Ca2+ pulse (“peak”), as in A. (C) Activation of RyR2 channel by luminal [Ca2+]. (A, Modified from Valdivia HH, Kaplan JH, Ellis-Davies GC, et al: Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science 267:1997–2000, 1995.) Regulation of RyR2 channels by luminal (intra-SR) Ca2+ has gained preeminence as a control mechanism of Ca2+ release, but this process remains as complex as cytosolic Ca2+ regulation. The crucial observation in ventricular myocytes is that, beyond a certain threshold, small changes in SR Ca2+ load result in far greater increases in Ca2+ release, with the relationship described as an inverse hyperbole with a high degree of cooperativity56 (see Figure 6-3). This is clearly an adaptive mechanism that promotes greater Ca2+ release (and stronger contractions) under conditions of increased Ca2+ uptake (such as occurs under β-adrenergic stimulation) and suggests that RyR2 channels increase their Po in response to elevated luminal [Ca2+]. The latter has been detected in single channel experiments,57,58 but the mechanism underlying luminal Ca2+ regulation at the molecular level remains uncertain and is possibly due to a combination of several of the following processes: (1) direct Ca2+ binding to RyR2 domains available only from inside the SR58,59; (2) activation of cytosolic sites by the Ca2+ ions being permeated by the channel (feed-through mechanism)60; and (3) binding of Ca2+ to calsequestrin and subsequent regulation of RyR2 through junctin, triadin, or both.47,61,62 In each of the approximately 10,000 individual couplons that occur in a typical ventricular myocyte, RyR2 channels are packed forming a paracrystalline array that promotes synchronized Ca2+ release.4,11 Each of these couplons is independently activated by Ca2+ entering the cell through the juxtaposed DHPRs of the T-tubules (junctional SR) or sarcolemma (peripheral couplings).1,4 The rapid spread of sarcolemmal depolarization into the interior of the cell by the T-tubules ensures that all individual couplons are simultaneously activated by ICa, generating a synchronized wave of Ca2+ release that spreads quickly to virtually all corners of the cardiomyocyte. In adult ventricular cells, ICa is insufficient to induce full contractions but is amplified threefold to eightfold (depending on the species) by Ca2+ release from the SR.1 Because Ca2+ is simultaneously the input and output signal of RyR2s, this amplification process (CICR) should intuitively be self-regenerating and all-or-none.63 In other words, the Ca2+ released by an RyR2 channel should activate further the same channel or its neighbors in an apparently interminable cycle. However, this is not observed experimentally; instead, CICR is finely graded by ICa and quickly terminates in intact cells. What counters the inherently positive feedback of CICR? At least three mechanisms have been proposed and, given the importance of CICR termination, it is likely that several mechanisms intervene (even redundantly) to avoid overflowing the cytosol with Ca2+. Ca2+-dependent inactivation of Ca2+ release was envisioned by Fabiato in his studies of CICR.3,8,52 The simplicity of this scheme makes it elegant. It involves two types of Ca2+ sites controlling the RyR2 channel: a fast-action, low-affinity Ca2+ activation site and a high-affinity but slow-action Ca2+ inactivation site.8 Accordingly, a fast Ca2+ stimulus evokes a transient burst of RyR2 activity because the channel is activated by Ca2+ acting on the activation site and then shut down as Ca2+ slowly binds to the inactivation site. On the other hand, a slow Ca2+ stimulus binds to the higher-affinity inactivation site and prevents channel openings, at least until the stimulus is removed. At sustained and high Ca2+ levels, estimated by Fabiato to be approximately 100 µM, the inactivation sites are saturated, leaving the channel in an absorbing inactivated state.52 Thus, this straightforward scheme allows for activation of Ca2+ release by the fast ICa and termination of CICR by the [Ca2+] lingering in the couplon after Ca2+ release. Recovery (repriming) of RyR2 channels from this inactivated (refractory) state requires removal of Ca2+ from the couplon, as expected to occur on a beat-to-beat basis. As intelligible as it sounds, this mechanism has encountered experimental and theoretical challenges. First, SR Ca2+ release was shown to be independent of the interval between two consecutive Ca2+ stimuli,64 which was not expected if RyR2 channels were refractory for a certain time after the first stimulus. In addition, when a sustained Ca2+ stimulus was used to inactivate the RyR2 channel, a subsequent Ca2+ stimulus reactivated Ca2+ release,65 which again was unexpected of a refractory process. Perhaps the most irreconcilable data come from single-channel experiments in which RyR2 channels inactivate at [Ca2+] >1 to 3 mM, which are levels unlikely to be reached during a normal contraction. Still, it is possible that at least some level of Ca2+-dependent inactivation of Ca2+ release occurs in vivo, as mathematical models estimate the [Ca2+] in the dyadic cleft at levels that are compatible with those required for partial inactivation of single RyR2 channels.66 In addition, RyR2 channels in situ may be more sensible to Ca2+ than those recorded under artificial environments. SR Ca2+ depletion has attracted attention as a mechanism to terminate CICR, but nagging issues remain. Although it is clear that RyRs terminate Ca2+ release once the Ca2+ inside the SR drops to a certain threshold (~50% of the total Ca2+ inside the store),67,68 the mechanisms underlying luminal Ca2+ regulation of RyRs remains unclear. In his classical studies that characterized calsequestrin (the major Ca2+-binding protein of the SR), Ikemoto et al.69 observed that the amount and the speed of Ca2+ release from SR vesicles depended directly on vesicular calsequestrin content rather than Ca2+ content, thus implying that calsequestrin “senses” the SR Ca2+ load and regulates the activity of RyRs. Boding well with this notion, Ca2+ sparks lasted longer or terminated prematurely after overexpression or partial depletion of calsequestrin levels in ventricular myocytes, respectively.70 Furthermore, purified RyR2 channels reconstituted in lipid bilayers exhibited little activation by luminal [Ca2+], but the addition of calsequestrin and its “anchors” junctin and triadin restored luminal Ca2+ sensitivity.57 Thus, the above data portrays calsequestrin as an indispensable component of the signaling mechanism that allows RyRs to terminate Ca2+ release upon [Ca2+]SR depletion; however, other data bestow little role on this protein. For example, RyR2 channels expressed in heterologous systems (and bearing no calsequestrin) exhibit robust luminal Ca2+ regulation and are activated by Ca2+ overload (store-overload induced Ca2+ release).58,59 In addition, in direct contradiction with some of these data, recombinant RyR2 channels (calsequestrin-free) reconstituted in lipid bilayers were activated by luminal [Ca2+],59 implying that luminal Ca2+ regulation is an intrinsic property of the channel protein. As a result, it is clear that RyRs are capable of sensing dropping levels of [Ca2+]SR to terminate Ca2+ release, but it is unclear whether they do it directly or through the calsequestrin-junctin-triadin ternary complex. As derangement of RyR refractoriness leads to Ca2+-dependent arrhythmias and may be involved in heart failure progression,71,72 luminal Ca2+ regulation of RyRs appears to be critical in the overall scheme to terminate CICR and likely depends on multiple factors. Stochastic attrition, the random closing of individual RyR2 channels, has been deduced from mathematical modeling as a potential mechanism to terminate CICR.63 High local [Ca2+] gradients build up in the couplon immediately right after CICR, but they quickly dissipate.66 If the number of RyR2 channels in a couplon was only one, it should be obvious that a random closure of that one RyR2 channel could be responsible for Ca2+ release termination (and dissipation of the [Ca2+] gradient). Stern63 inferred that stochastic attrition could terminate local Ca2+ release if the number of RyR2 channels in a couplon was fewer than 10, but higher number of channels would decrease its probability precipitously. Because ultrastructural and functional data estimate this number to be close to 100,1,18
Structural and Molecular Bases of Sarcoplasmic Reticulum Ion Channel Function
The Sarcoplasmic Reticulum
Structural Arrangement of the Sarcoplasmic Reticulum
Molecular Players of Excitation-Contraction Coupling
Methods of Recording Sarcoplasmic Reticulum Ion Channels
Reconstitution of Sarcoplasmic Reticulum Channels in Lipid Bilayers
Confocal Imaging of RyR Ca2+ Fluxes
[3H]Ryanodine Binding As an Index of RyR Activity
Electron Probe X-Ray Microanalysis of Ca2+ and Other Ions Within the Sarcoplasmic Reticulum
Molecular Structure of the Cardiac Ryanodine Receptor (RyR2)
Accessory Proteins of the RyR2 Channel
Ca2+ Regulation of RyR2 Channels
Regulation of Isolated RyR2 channels
Regulation of RyR2 Channels In Situ
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Structural and Molecular Bases of Sarcoplasmic Reticulum Ion Channel Function
