7 Mitochondria are the power plants of all kinds of tissues/cells. In heart/cardiomyocytes, mitochondria use glucose and fatty acids to produce adenosine triphosphate (ATP) that drives muscle contraction and relaxation for pumping blood that circulates through the entire body during each heart beat unceasingly, up to a centenary human life. Indeed, mitochondria were originally found and studied mostly as a cellular “powerhouse” in the first half of the 20th century. Soon it was also recognized that Ca2+ stimulates oxidative phosphorylation (OXPHOS) and electron transport chain (ETC) activity, which results in the stimulation of ATP synthesis1 (Figure 7-1, A, B). Early studies in the 1960s to 1970s revealed that isolated mitochondria can take up a large quantity of Ca2+. It is surprising to note that super-physiological high Ca2+ concentrations ([Ca2+]) (10 to 100 µM) were required to activate Ca2+ uptake into isolated mitochondria (see reviews2–4). However, in the intact cells, less than a 10-µM [Ca2+] global increase in the cytosol propagated into the mitochondria matrix. This discrepancy between isolated mitochondria and intact cells was partially resolved by the finding of high cytosolic [Ca2+] ([Ca2+]c) at the microdomains between mitochondria and endoplasmic reticulum (ER)/sarcoplasmic reticulum (SR) during ER/SR Ca2+ release via inositol triphosphate (IP3) receptor and/or ryanodine receptor (RyR), as a result of the juxtaposition of these two organelles. These seminal discoveries have finally positioned mitochondria as one of the key players in the dynamic regulation of physiological Ca2+ signaling and have promoted research in the field of mitochondrial Ca2+ channels/transporters. Soon it was also discovered that a Ca2+ efflux mechanism exists to dump the accumulated matrix Ca2+ into the cytosol. Although the functional characteristics of mitochondrial Ca2+ influx/efflux mechanisms were functionally discovered more than 50 years ago, the molecular identities responsible for these mechanisms have remained a mystery until very recently. The mitochondrial Ca2+ influx was dogmatically considered to result from a single transport mechanism mediated by a mitochondrial Ca2+ uniporter (MCU) principally as the result of nearly complete inhibition by ruthenium red (RuR) and lanthanides, and its channel nature was originally proposed more than 30 years ago.2,4 However, subsequent studies have identified additional Ca2+ uptake pathways, which exhibit different Ca2+ affinity, uptake kinetics, and pharmacological characteristics from the original MCU theory (see review3) (Figure 7-1, C). These distinct properties are critical for enabling mitochondria to carry out multiple Ca2+-mediated functions with optimal spatial and temporal effectiveness. Among these studies, RyR1 was found as the first mitochondrial Ca2+ influx mechanism with a known molecular identity reported from our group (Figure 7-1, C). IMiCa was recently recorded from mitoplasts (used mitochondria without the outer mitochondrial membrane [OMM]),5 providing direct electrophysiological evidence for the existence of a Ca2+ selective ion channel, which would possibly fit he originally predicted channel nature of MCU. Through RNA interference studies, several groups have recently proposed novel candidate proteins that are involved in mitochondrial Ca2+ uptake mechanisms such as leucine zipper-EF-hand containing transmembrane protein 1 (Letm1)6 and mitochondrial calcium update (MICU1),7 in addition to our RyR1 discovery (Figure 7-1, C). Finally, two very recent articles reported that the coiled-coil domain containing protein 109A (CCDC109A) is the MCU molecular identity.8,9 Figure 7-1 Overview of Mitochondrial Ion Channels/Transporters In addition to Ca2+, the movements of various electrolytes and metabolites across the inner mitochondrial membrane (IMM) as well as the OMM are important for the regulation of major mitochondrial functions, including ATP synthesis, Ca2+ homeostasis, and reactive oxygen species (ROS) and nitrogen species (RNS) generation. Unlike other organelles, mitochondria possess unique double-membrane structures with distinctive phospholipids and protein compositions, which allow mitochondrial membranes to maintain a mitochondrial membrane potential (Δψm) and unique architecture including cristae (see next section). It is interesting to note that IMM and OMM have different sets of ion channels/transporters, as summarized in Figure 7-1A. These include (1) proton (H+) movement related to ETC activity and uncoupling proteins (UCPs) for the maintenance of Δψm at IMM; (2) K+-selective and anion-selective pathways at IMM and across the two membranes, such as the mitochondrial permeability transition pore (mPTP), which are important for the maintenance of mitochondrial volume; (3) the movement of metabolites including ATP, adenosine diphosphate (ADP), and phosphate (Pi) at IMM and OMM, and (4) release of proapoptotic proteins, which potentially leads to cell death mainly at OMM (see III) or mPTP. In this chapter, we summarize recent progress in mitochondrial ion channel/transporter research; this is followed by an overview of cardiac mitochondrial ion channel/transporter biophysics and cardiac physiology and pathophysiology. The most prominent contribution of mitochondria to cellular metabolism is based on their capacity to generate ATP through the tricarboxylic acid (TCA) cycle and OXPHOS through the ETC, which is a concerted series of redox reactions catalyzed by four multi-subunit enzymes embedded in the IMM (complexes I-IV) and two soluble factors, cytochrome c (cyt c) and coenzyme Q10 (Co Q10), which function as electron shuttles within the mitochondrial intermembrane space (IMS) (see Figure 7-1, B) (see I.A and II.D). In a healthy eukaryotic cell, more than 90% of the total intracellular ATP is generated by mitochondria. The main driving force of OXPHOS is known as “chemiosmosis,” which is generated by proton (H+) movement across IMM that creates a membrane potential (Δψm, negative in the matrix) and a pH gradient (ΔpH, alkaline in the matrix). Chemiosmosis is the movement of ions across a selectively permeable membrane, down their electrochemical gradient (protonmotive force: Δp), which is determined by both Δψm and ΔpH components across the IMM (Δp =Δψm + ΔpH). The chemiosmotic hypothesis was first proposed by Peter D. Mitchell in 1961.10 The basic assumption of this chemiosmotic theory is derived from the important observation that the IMM is generally impermeable to ions, but it keeps the permeability of H+. The composition of the OMM is similar to those of sarcolemma (SL) and ER/SR in eukaryotic cells, whereas the IMM does not possess cholesterol but has a unique dimeric phospholipid, cardiolipin, which is a typical composition for bacterial membranes. Cardiolipin has a unique ability to interact with proteins, including several mitochondrial respiratory chain complexes (I, III, IV, and V), and support their activities11 while also contributing to maintaining the structure of cristae, which enhances the efficiency of ETC activity, possibly through facilitating the formation of super-complexes of respiratory chain at the IMM.12 The unique structure of cardiolipin serves as an H+ trap at the IMS near the IMM, maintains the pH change near the IMM, and efficiently pools H+ or releases H+ to the mitochondrial ATP-synthase (complex V) at IMM (Figure 7-1, B).10,11 It is interesting to note that the IMM and the OMM not only have different phospholipid compositions, but they show different protein-to-lipid ratios (for OMM, about 0.5 : 0.5; and for IMM, about 0.8 : 0.2). This may allow the proteins at the IMM to possess enzymatic and/or transport functions compared with those at the OMM, thus the IMM is much less permeable to ions and small molecules than is the OMM, which also provides the cellular compartmentalization between the mitochondrial matrix and cytosol. As is shown in Figure 7-1, B, complexes I, III, and IV are engaging with the translocation of H+ from matrix to IMS, which establishes Δψm and ΔpH, as well as Δp. Therefore, Δψm is usually highly negative (around −180 to −190 mV) compared with the resting potential at plasma membranes. Finally, according to the mentioned mechanisms, this large driving force for H+ influx (Δp) is used by complex V to produce ATP (see Figure 7-1, B). Other important roles for Δp in addition to ATP synthase at the IMM are that (1) ΔpH drives pyruvate transport through pyruvate carrier (PYRC) into the matrix; (2) ΔpH drives Pi transport through Pi carrier (PIC) into the matrix; and (3) Δψm drives ATP/ADP exchange through the adenine nucleotide translocator (ANT) (see Figure 7-1, A, B). As mentioned above, Ca2+ uptake into the mitochondrial matrix stimulates ATP synthesis (see Figure 7-1A). At the resting state, the electrochemical driving force for Ca2+ uptake is also provided by Δψm across the IMM. For MCU, Ca2+ is taken into the mitochondrial matrix down its electrochemical gradient without transport of another ion. Basically, for each Ca2+ transported through MCU, there is a net transfer of two positive charges into matrix, resulting in a drop in Δψm, which is energetically unfavorable. However, the Ca2+-stimulated respiration not only will compensate the loss of Δψm by the efflux of H+ through ETC, it will also produce a net gain of ATP. In addition, multiple Ca2+ efflux mechanisms work in concert to expedite a transient and an oscillatory nature rather than a tonic and a steady state change of matrix [Ca2+] ([Ca2+]m). Superoxide and nitric oxide are ROS and RNS, respectively, produced in cells under normal physiological conditions; both species react with other molecules and with each other to form a diverse array of additional ROS and RNS.13 High levels of ROS and RNS are known to promote cell damage and death, but the production of low to moderate levels of ROS/RNS is critical for the proper regulation of many essential cellular processes, including gene expression, signal transduction, and cardiac excitation-contraction (E-C) coupling.14 ROS are generated by several different cellular sources: (1) membrane-associated nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, (2) cytosolic xanthine and xanthine oxidase, and (3) the mitochondrial ETCs at IMMs. Superoxide is the primary oxygen free radical produced in mitochondria via the slippage of an electron from the ETC to molecular oxygen during OXPHOS (see Figure 7-1, B). This “constitutive” superoxide generation is central to proper cellular redox regulation. Recent studies from our collaborating groups detected a “stochastic” and “transient” superoxide burst from either single or restricted clusters of interconnected mitochondria across a wide variety of cell types, termed a mitochondrial superoxide flash (mSOF).15 A transient depolarization of Δψm is associated with each mSOF. The proposed mechanism of mSOF in cardiac muscle cells is as follows: A small increase in constitutive ROS production transiently opens a large channel named the mPTP to cause Δψm depolarization, which subsequently stimulates the ETC to produce a burst of superoxide production. This idea is similar to the previous observation that the mPTP opens and closes transiently (“flicker”) at its low conductance state and can release Ca2+ from matrix2. The frequency of mSOFs varies widely across different cell types and experimental conditions (disease models), suggesting that these flashes act as biomarkers of cellular metabolic activity and oxidative condition in physiological and pathophysiological conditions.16 Future studies will clarify the contribution of altered mSOF activity to ROS overproduction and metabolic dysfunction in a wide range of mitochondrial diseases and oxidative stress–related disorders. Finally, it should be noted that there is controversy surrounding whether the mSOF might reflect oscillations in mitochondrial pH.17 Based on chemiosmotic theory, one can envision that whenever a significant drop in Δψm occurs, there should be an alkalization in ΔpH. Thus, the interrelationship between mSOF, pH transients, and Δψm fluctuations will be an important research topic in the future. Programmed cell death is genetically designed for self-killing of individual cells and is one of the critical mechanisms for maintaining homeostasis of multicellular organs/tissues, including the heart.18 Apoptosis is a well-established mechanism of programmed cell death activated by a variety of cellular stresses and signals.19 In mammalian cells, the activation of caspases (a family of cysteine proteases) is the central mechanism for apoptosis. Under the resting condition, caspases are tightly kept inactive as a “proenzyme” form and/or by binding to inhibitory proteins (named inhibitors of apoptosis) in the cytosol. During apoptosis, caspases are activated by cleavage that changes the “proenzyme” form to the enzyme form and by dissociation of the inhibitors of apoptosis.20 One of the major apoptotic pathways is derived from mitochondria, and this contributes to the activation of caspases in the cytosol. This mechanism is initiated by the release of proapoptotic factors from mitochondria, including cyt c, Smac/Diablo, and HtrA2/Omi. Cyt c released to cytosol activates caspases through its binding to apoptotic protease activating factor 1 (Apaf-1). Smac/Diablo and HtrA2/Omi bind to the inhibitory proteins to remove their inhibitory effects from caspases. The apoptotic DNases, including apoptosis inducing factor (AIF) and endonuclease G (Endo G), are also released from mitochondria, as are the aforementioned proapoptotic factors. The release of these proapoptotic factors is regulated under the tight control of mitochondrial membrane permeability.21 This mitochondrial membrane permeabilization is mediated via at least three distinct mechanisms: (1) physical rupture of the OMM as a result of mitochondrial swelling (usually linked to mPTP opening) (II.B.2), (2) modification of the structure of the voltage-dependent anion channel (VDAC) through interaction with proapoptotic proteins such as the Bcl-2 protein family, and (3) formation of a new pore as a consequence of oligomerization and membrane insertion of proapoptotic proteins, including the Bcl-2 family of proteins. The first pathway involves Δψm depolarization and swelling of the matrix space, followed by loss of OMM integrity and rupture spilling of proapoptotic proteins into the cytosol. In contrast, the other two pathways occur without Δψm depolarization. At first, an important regulatory protein of the mitochondrial Ca2+ uptake mechanism was identified using bioinformatics and siRNA screening, termed MICU17. MICU1 has two Ca2+-binding EF-hands, but has only one single membrane-spanning domain, which seems unlikely to form a channel pore and to be an MCU itself rather than a modulator of MCU (see Figure 7-1, C). Finally, two recent articles from two different groups simultaneously reported CCDC109A as the molecular identity of the MCU.8,9 The main characteristics of the MCU are as follows: (1) CCDC109A has two transmembrane domains, which seem likely to make a channel pore; (2) knock-down of CCDC109A dramatically reduces mitochondrial Ca2+ uptake, and this effect was rescued by overexpression of MCU; (3) CCDC109A knock-down itself does not affect mitochondrial O2 consumption, ATP synthesis, Δψm, and morphology, even though Ca2+ influxes are critical for regulating these functions; (4) reconstituted MCU in lipid bilayers shows RuR-sensitive Ca2+ current with 6-7 pS single-channel activity9; and (5) site-specific mutations at the MCU pore region show loss of function. Regarding the structure of MCU, the topology of MCU is still under debate. CCDC109A seems to possess two transmembrane domains that oligomerize to become a Ca2+ channel. Rizzuto’s group proposed that C- and N-terminals face to the IMS, whereas Mootha’s group proposed that they face the opposite direction.22 The discrepancy in the topology of MCU will need to be resolved for an understanding of the modulation of MCU functions by signaling molecule from the cytosol or from the matrix. One of the candidates for the mitochondrial Ca2+ uptake mechanism with a known molecular identity is the mitochondrial RyR in cardiac cells reported from our group.23 There are three different RyR isoforms (RyR1, 2, and 3) with different physiological and pharmacological properties. In cardiac cells, intracellular Ca2+ release during E-C coupling was mainly controlled by RyR2 located in the SR (see Figures 7-1, A-C). Although RyR1 is also detectable at mRNA and protein levels in cardiac tissue, its functional and physiological roles in the heart have not been fully understood for a long time. We first showed that a low level of functional RyR is also expressed at the heart IMM, and this has a role in the fast Ca2+ uptake pathway24 (see Figures 7-1, B and 7-2). Furthermore, RyR in cardiac mitochondria exhibits remarkably similar biochemical, pharmacologic, and functional properties to those of RyR1 in skeletal muscle SR, but not to those of RyR2 in cardiac SR. Therefore, we termed this as mRyR1 (mitochondrial RyR1)(see review3). The molecular identity of mRyR1 was carefully analyzed and confirmed by a variety of functional and biochemical experiments using not only native heart, but also the RyR1 knockout mouse heart. Recent studies from our lab using electrophysiological techniques directly demonstrated the existence of mRyR1 and clearly showed the predicted channel nature of skeletal RyR1. In a conventional lipid bilayer of mRyRs purified from a heart IMM, the activity of RyR1 but not of RyR2 was observed.25 The biophysical and pharmacologic properties of native single mRyR1 channels were further characterized in heart mitoplasts using patch-clamp techniques.26 We observed a novel 225-pS cation-selective channel in heart mitoplasts that exhibited multiple subconductance states, which was blocked by high concentrations of ryanodine and RuR, the known inhibitors of RyRs. Ryanodine exhibited a concentration-dependent modulation of this channel, with low concentrations stabilizing a subconductance state and with high concentrations abolishing activity. The channel properties of Ca2+-dependent [3H]ryanodine binding and the channel modulation by caffeine in isolated cardiac mitochondria24 are suggesting that the topology of mRyR1 is the same as that of RyR1 at SR because of these agonist-binding sites; C- and N-terminals face to the IMS (corresponding to the cytoplasmic side of RyR1 at SR), and S1-S2, S3-S4, and S5-S6 linkers face to the mitochondrial matrix side (corresponding to the SR luminal side of RyR1 at SR). However, further studies will be needed to confirm the topology of mRyR1 using other modulators from both the matrix side and the cytosolic side. Figure 7-2 Activation/Deactivation Patterns of Ca2+-Influx/Efflux Mechanisms Unlike MCU, RyR is a poorly Ca2+-selective, large cation channel.26 Therefore, opening of mRyR1 might collapse Δψm, which is energetically unfavorable. This dichotomy would be explained as follows: (1) The expression number of RyR1 in a single mitochondrion is very small, and the changes in Δψm might be minimized locally, ensuring maintenance of a Ca2+-driving force; (2) the rapid Ca2+-activation and -inactivation profile of this channel (see Figure 7-2) would minimize the Δψm change instantly; and (3) any small decrease in Δψm can be readily compensated by the Ca2+-dependent activation of dehydrogenase during the TCA cycle and ATP synthesis. Taken together, the mRyR may be uniquely poised to sequester Ca2+ during a transient and rapid excitation-contraction coupling process in cardiac muscle cells.
Structural and Molecular Bases of Mitochondrial Ion Channel Function
Historical Overview of Mitochondrial Ion Channel/Transporter Research
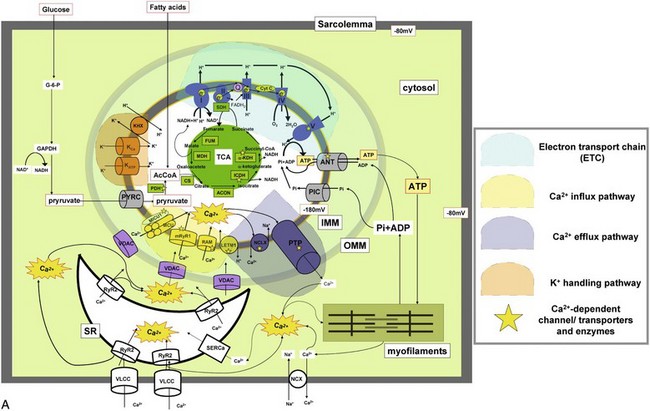
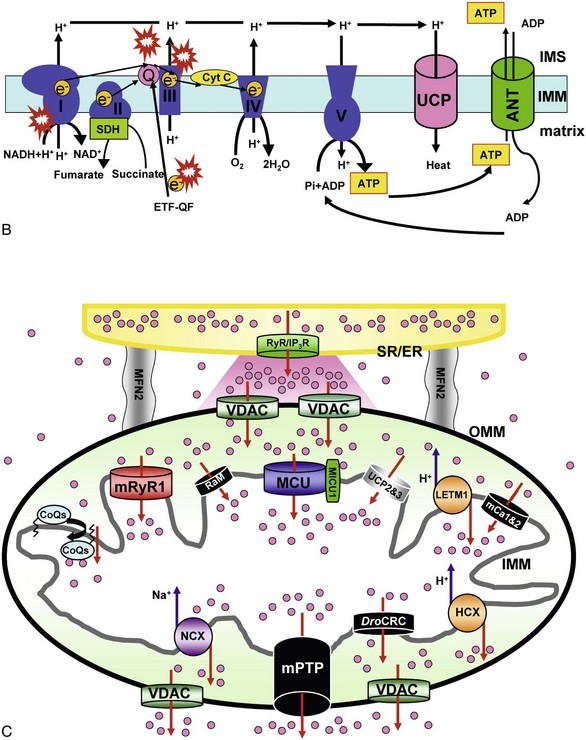
A, Major ion movements in mitochondria. Ca2+-dependent ion channels/transporters and enzymes are indicated with stars. B, Mitochondrial electron transport chain (ETC) and possible sites of superoxide production. Red “explosion” symbols indicate places where superoxide production occurs. ETF-QF, electron transferring flavoprotein-quinone oxidoreductase; Q, coenzyme Q10. C, Ca2+-influx/efflux mechanisms: The channels/transporters for which molecular identities are still unknown are shown as black. Red arrows show Ca2+ movements, and blue arrows show other ion movements. DroCRC, Ca2+-release channel in Drosophila mitochondria; RaM, the rapid mode of uptake. (©O-Uchi J et al: Perspectives on: SGP symposium on mitochondrial physiology and medicine: Molecular identities of mitochondrial Ca2+ influx mechanism: Updated passwords for accessing mitochondrial Ca2+-linked health and disease. J Gen Physiol 139:435–443, 2012. Originally published in Journal of General Physiology, doi:10.1085/jgp.201210795.)
Overview of Mitochondrial Bioenergetics and Mitochondrial Membrane Potential
Overview of Mitochondrial ROS Generation and Mitochondrial Membrane Potential
Overview of Mitochondria-Induced Apoptosis and Mitochondrial Ion Channels
Ion Channels/Transporters at Mitochondrial Inner Membrane
Mitochondrial Ca2+ Channels/Transporters Regulating Mitochondrial Ca2+ Influx
Mitochondrial Ca2+ Uniporter (MCU)
Mitochondrial Ryanodine Receptor (mRyR)
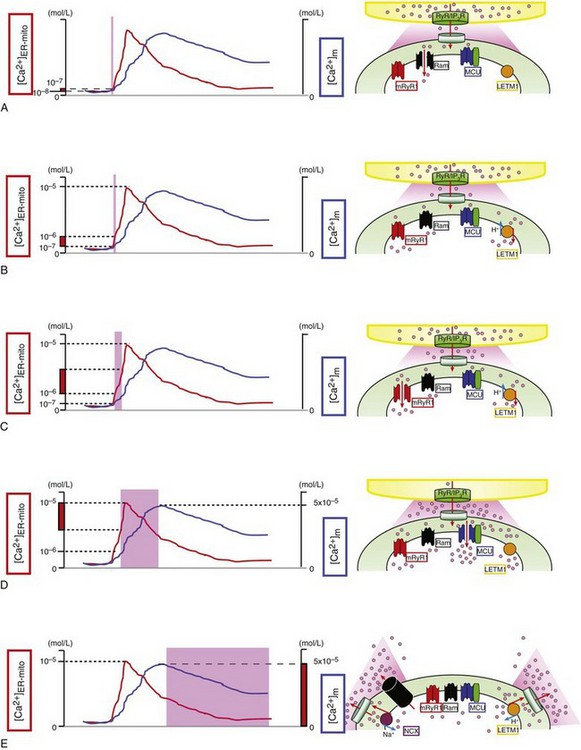
A, At first, the rapid mode of uptake (RaM) (black) is activated at the very initial phase of Ca2+ transient at microdomains between mitochondria and ER/SR ([Ca2+]ER-mito) transient (red line) (<200 nM) with faster Ca2+ uptake kinetics (ms time scale). B, Letm1 (orange) starts to uptake Ca2+ at ≥200 nM [Ca2+]ER-mito. C, mRyR1 (red) starts open at ≈1 µM [Ca2+]ER-mito with a 5-fold faster Ca2+ transport compared with the MCU, and deactivates before [Ca2+]ER-mito reaches the peak. D, Finally, MCU (blue) starts to activate at >1 µM [Ca2+]ER-mito, and the activity increases in a [Ca2+]ER-mito,-dependent manner. At this point, Letm1 (orange) shifts from Ca2+-uptake mode to Ca2+-efflux mode. E, mPTP (black) and NCX (purple) contribute to Ca2+ efflux in mammalian cells and form the decay phase of [Ca2+]m transient (blue line). Letm1 also works as a Ca2+-efflux pathway at this phase. (©O-Uchi J et al: Perspectives on: SGP symposium on mitochondrial physiology and medicine: Molecular identities of mitochondrial Ca2+ influx mechanism: Updated passwords for accessing mitochondrial Ca2+-linked health and disease. J Gen Physiol 139:435–443, 2012. Originally published in Journal of General Physiology. doi:10.1085/jgp.201210795.)



