Fig. 7.1
Human sternal development . Development continues after birth until somatic growth is completed [from van der Merwe, A.E., et al., A review of the embryological development and associated developmental abnormalities of the sternum in the light of a rare palaeopathological case of sternal clefting. Homo, 2013. 64(2): p. 129–41]
During normal growth and development, the primary ossification centers enlarge and fuse to form the adult sternal body while the superior most ossification center forms the manubrium. This process is completed once somatic growth is complete. At various time points during development this process can be disturbed either due to arrest of development or environmental factors that alter maturation.
Failure of fusion or failure in the formation of the sternal bars results in a sternal defect. These defects may occur anywhere along the sternum. Small areas within the sternum where there is a failure of fusion form sternal foramina, if they extend from superior or inferior or if there is complete failure to fuse they form sternal clefts. The superior sternal cleft is thought to be due primarily to lack of development of the manubrium rather than lack of fusion of the sternal bars. Between 30 and 33 weeks gestation the four or five ossification centers can be seen by ultrasound and sternal clefts can be identified at this time [3].
Associated Conditions
Sternal defects , while rare, have a strong association with life-threatening congenital deformities of thoracic and abdominal organs. The presence of a sternal defect should prompt a thorough evaluation of the thoracic cavity, abdomen, and a genetic evaluation. A broad spectrum of deformities have been described involving the sternum, heart, vasculature, and upper abdominal wall. Cardiac anomalies may include ventricular septal defect, double-outlet right ventricle, valvular pathology, and conotruncal anomalies such as Tetralogy of Fallot. Hypoplastic left heart syndrome has been described as well. An association between midline defects and cardiac diseases was made in the 1950’s. This was first described by Cantrell in 1958 with findings including: a midline supra-umbilical thoraco-abdominal wall defect, a lower sternal defect, a diaphragmatic pericardial defect, a deficiency of anterior diaphragm and, various intracardiac anomalies [4]. We now refer to this group of defects as Pentalogy of Cantrell. The proposed mechanism is a failure of the lateral mesodermal folds to migrate to the midline, causing the sternal and abdominal defects along with defects in the anterior diaphragm and pericardium. Sternal and abdominal wall defects cause herniation of organs, leading to ectopia cordis and omphalocele. The gene or genes responsible for this have been mapped to the X chromosome (Xq25-Xq26). The syndrome has an estimated incidence of 5.5 per one million live births [5]. Diagnoses of complete pentalogy of Cantrell are extremely rare. Most diagnosed cases are classified as incomplete variants with the subjects meeting three or four out of the five criteria [6]. In addition, some cases describe an association of sternal malformation with other vascular anomalies and vascular dysplasia such as giant ascending aortic aneurysm, aortic coarctation, and coronary ostial abnormalities [7]. It may be that the mesoderm that is important for normal chest wall development also plays a role in development of the conotruncus.
Sternal defects can also be associated with superficial craniofacial vascular lesions. Clinical features associated with this condition include cutaneous craniofacial hemangiomas, sternal cleft, atrophic abdominal raphe extending from the sternal defect to the umbilicus, and associated vascular malformations on internal organs such as the respiratory tract and visceral organs [8]. This spectrum may represent part of the PHACES (Posterior fossa malformations–hemangiomas–arterial anomalies–cardiac defects–eye abnormalities–sternal cleft and supraumbilical raphe) syndrome. Children with sternal malformation and hemangiomas should be carefully evaluated for cardiac, ophthalmologic, and neurologic malformations. The syndrome has an X-linked inheritance pattern and is therefore much more common in females than in males (8:1).
Diagnosis
The diagnosis of sternal malformation is easily done at birth by physical exam. The most striking finding is one of bulging of the skin overlying a partial sternal cleft or in the case of complete sternal cleft, the precordial pulse. In inferior sternal clefts often associated conditions as mentioned earlier can be seen. Prenatal diagnosis is possible. As previously mentioned, sternal defects are associated with other life-threatening malformations, thus diagnostic investigations have been directed to exclude those associated anomalies. Ultrasonography has been widely used to assess fetal development and has been reported to be of use in early diagnosis. The use of two-dimensional and three-dimensional ultrasonography has been used for early diagnosis and referral to specialized centers [9, 10].
Sternal Clefts
The exact incidence of sternal malformation is unknown, but it seems to have a 2:1 female predominance [11]. In most cases, infants with sternal cleft are asymptomatic and surgical repair can be performed to provide protective coverage of the underlying heart. The most common of this group of congenital chest wall malformations, this arises from failure of the embryonic fusion of the sternal bars. The defect can be either complete or partial. In a recent review of congenital sternal anomalies , 67 % were superior clefts, 19.5 % complete, 11 % inferior, and 2.5 % were sternal foramen [11]. Sternal clefts are often seen as part of other rare syndromes such as PHACES, Cantrell’s pentalogy, sternal malformation/vascular dysplasia, and other midline defects [11]. See Fig. 7.2.
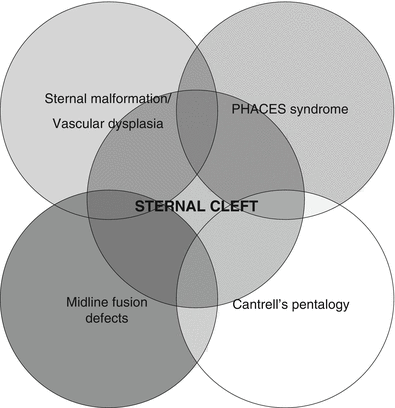
Fig. 7.2
Rare associations with sternal cleft [From Torre, M., et al., Phenotypic spectrum and management of sternal cleft: literature review and presentation of a new series. Eur J Cardiothorac Surg, 2012. 41(1): p. 4–9]
Surgical repair of sternal clefts is indicated to protect the thoracic or abdominal viscera. There have been many different approaches to closure described in the literature [11] and this speaks to the variability in presentation and in anatomy that is seen with these lesions. Repair with autologous tissue and with prosthetic material has been reported with good results although primary closure and repair is preferable if the patient will tolerate this. See Table 7.1.
Surgical procedure | n/70 | % | Gaslini (n/7) | % |
|---|---|---|---|---|
Primary closure alone | 27 | 38.6 | 4 | 57.14 |
Primary closure with periosteal flap | 14 | 20.0 | – | – |
Primary closure with sliding chondrotomies | 10 | 14.3 | – | – |
Primary closure with cartilage resection | 4 | 5.7 | – | – |
Bone graft placement | 7 | 10.0 | – | – |
Prosthetic closure | 5 | 7.1 | 3 | 42.86 |
Muscle flap | 2 | 2.9 | – | – |
Two-stage primary closure | 1 | 1.4 | – | – |
Ectopia Cordis
In this condition, the heart is malpositioned outside of the thoracic cavity and not covered by skin or other somatic structures, known classically as “the naked heart.” It was first reported in 1671 by Stensen. The first reported successful repair was at the Children’s Hospital of Philadelphia by Koop in 1975. As of 1995 the patient was reported to be alive and well but required multiple reoperations due to the prosthetic material used to reconstruct the anterior chest wall. The apex of the heart is often oriented in the anterior cephalad position and usually has intrinsic anomalies. The sternal defect can be superior, inferior, or total. On rare occasions, the heart may protrude through a defect in the central portion of the sternum. The lack of overlying somatic tissue , along with a hypoplastic thoracic cavity, makes surgical correction very difficult. Isolated survival has been reported in several cases after staged surgeries [12]. See Fig. 7.3. Complicating the management of these patients is the rarity of the lesion combined with the timeliness of surgery that these patients require to survive. Typically these patients do not tolerate repositioning of the heart within the hypoplastic thorax and this is what leads to death. There are numerous different types of ventral wall defects with ectopia cordis [13, 14]. See Table 7.2.

Fig. 7.3
Total thoracic ectopia cordis [From Alphonso, N., et al., Complete thoracic ectopia cordis. Eur J Cardiothorac Surg, 2003. 23(3): p. 426–8]
Sternal defect | No. of cases | Abdominal wall defect | No. of cases | Not reported |
|---|---|---|---|---|
Completely absent | 10 | Omphalocele | 40 | |
Xyphoid defect | 10 | Diastasis recti | 15 | |
Manubrial defect | 3 | Eventration | 8 | |
Defect of body of sternum | 10 | Umbilical hernia | 3 | |
Partial defect | 2 | |||
One-third lower sternum defect | 24 | |||
Two-thirds lower sternum | 11 | |||
Bifid sternum | 10 | |||
Total | 80 | Total | 66 | 71 |
Cervical Ectopia Cordis
This is an extremely rare condition in which the heart is displaced cranially, sometimes with the cardiac apex fused with the mouth. These patients usually have severe craniofacial anomalies and often have bands and other anomalies that are not compatible with survival. Prognosis is always poor and there have been no reports of survivors to date.
Thoraco-Abdominal Ectopia Cordis
The heart is covered by a thin membranous or cutaneous layer that is often pigmented. An inferior sternal cleft is present and the heart can be located within the thorax or be displaced into the abdomen. There are often intrinsic cardiac anomalies present, but there is no cardiac malrotation [15]. This malformation is often found as part of the rare syndrome known as the pentalogy of Cantrell. Cantrell defined this syndrome as having five characteristic findings including omphalocele, anterior diaphragmatic hernia, sternal cleft, ectopia cordis, and an intracardiac defect [4]. Successful repair and long-term survival are much more frequent in thoraco-abdominal ectopia cordis than thoracic ectopia cordis. Ventricular diverticula can also be seen in this condition and are associated with omphalocele and sternal cleft [16]. See Fig. 7.4.
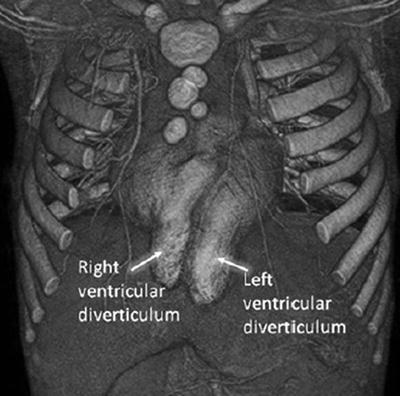
Fig. 7.4
Ventricular diverticula with Pentalogy of Cantrell [Nagashima, M., T. Higaki, and A. Kurata, Ectopia cordis with right and left ventricular diverticula. Heart, 2010. 96(12): p. 973]
Surgical repair includes reconstruction of the ventral thoracic and abdominal wall as well as diaphragm. Any congenital heart disease present is typically repaired at the time of surgery.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


