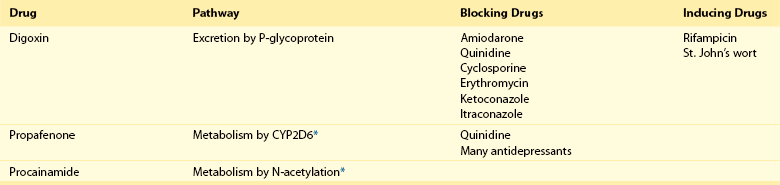
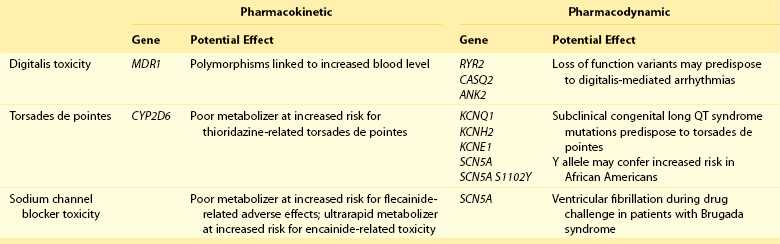
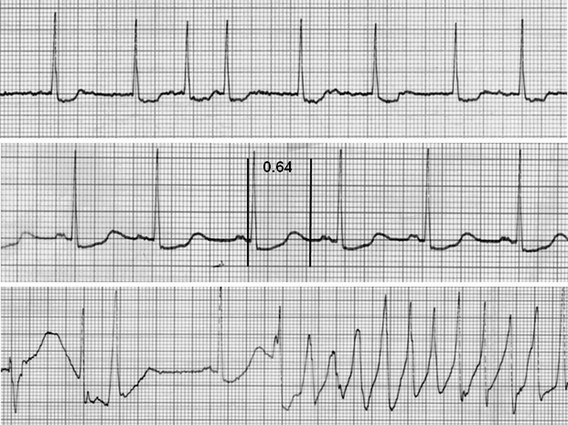
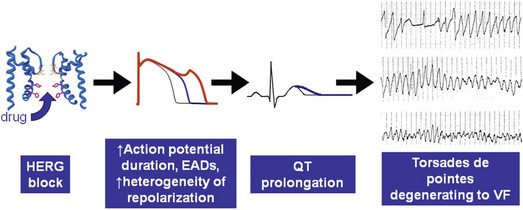
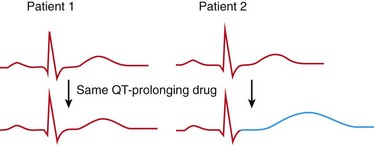
111 Cardiac arrhythmias remain a major source of morbidity and mortality in developed countries. In the past decade, the treatment of arrhythmias has been altered dramatically by the development of nonpharmacologic therapies, such as targeted ablation of arrhythmogenic tissues and implantable cardioverter defibrillators (ICDs), as well as the limited efficacy and proarrhythmic potential of conventional antiarrhythmic drugs. Efficacy of antiarrhythmic drugs in suppressing the most common arrhythmia—atrial fibrillation (AF)—is generally 30% to 60% and no single agent shows superiority with the possible exception of amiodarone. Between 500,000 and 1 million North Americans and Europeans die each year of sudden cardiac death (SCD), which causes 10% to 20% of all deaths among adults in the Western world.1,2 The most common underlying cause for SCD is ventricular fibrillation (VF). SCD most often occurs in patients with underlying coronary artery disease (CAD). Therefore, as the prevalence of CAD increases worldwide, so will the incidence of SCD. Although identification of patients at high risk for SCD is possible by evaluating those with heart disease, SCD is the initial presenting symptom of heart disease in approximately half of all cases.3 ICDs are often used in those judged to be at high risk of SCD. These devices are highly effective at terminating life-threatening arrhythmias if they occur, but they do not prevent arrhythmia. The Cardiac Arrhythmia Suppression Trial (CAST) represents a landmark trial result, not only for antiarrhythmic drug development but for drug development in general.4 The CAST tested the prevalent hypothesis that because ventricular ectopy following myocardial infarction (MI) is a risk factor for SCD, suppression of ventricular ectopy would reduce the incidence of SCD. Mortality among patients randomized to ventricular ectopic suppression with encainide or flecainide in the CAST was approximately threefold higher than that of patients randomized to receive placebo. The study also emphasized the importance of a randomized, placebo-controlled trial with a primary “hard” endpoint, such as death, as opposed to a surrogate end point, such as ectopic beat suppression, to determine whether a drug was beneficial. The drugs tested in CAST were potent sodium channel–blocking agents. For basic and clinical scientists interested in arrhythmia mechanisms, the CAST result provided a strong impetus to further work that defined the way in which loss of sodium channel function was arrhythmogenic. It is thought that blocking sodium channels increases the risk of SCD by slowing conduction or increasing the heterogeneity of repolarization, both of which can be arrhythmogenic, especially in diseased hearts.5,6 Multiple lines of evidence support the hypothesis that loss of sodium current (INa) slows cardiac conduction and may be arrhythmogenic; such loss can be structural (e.g., mutations in the cardiac sodium channel gene SCN5A leading to loss of peak INa),7–9 pharmacologic,7,10 or reflect activation of second messenger systems. For example, protein kinase A (PKA) activation has been reported to increase peak INa, at least in part because of altered trafficking,11 whereas protein kinase C activation decreases it.12 Another consequence of CAST was that development of sodium channel–blocking drugs came to a rapid halt and QT-prolonging agents then assumed the limelight. Just as procainamide and lidocaine provided the structural starting point of a range of sodium channel–blocking molecules, two compounds with prominent action potential (AP)-prolonging properties formed the framework here: N-acetyl procainamide (NAPA) and sotalol. Both compounds also did not block the sodium channel and gave rise to a generation of QT-prolonging drugs, such as d-sotalol, dofetilide, almokalant, and sematilide. Although the screening assay to develop these agents usually was APD in a guinea pig papillary muscle, and as with newer sodium channel blockers, these compounds were not synthesized to interact with a specific predefined molecular target. Nevertheless, subsequent studies identified block of one specific potassium current, termed IKr (the rapid component of the cardiac delayed rectifier potassium current), as the major mechanism underlying QT prolongation by antiarrhythmic drugs. IKr is encoded by KCNH2 (also known as HERG). However, blocking IKr carries the risk of marked QT interval prolongation and a distinctive form of polymorphic ventricular tachycardia, termed torsades de pointes. Indeed, potassium-channel blocking antiarrhythmic drugs have been tested in CAST-like trials and did not prevent more deaths than did placebo.13–15 Moreover, the same mechanism underlies the development of torsades de pointes in response to noncardiovascular drugs, such as certain antihistamines, antibiotics, and antipsychotics.16 The term antiarrhythmic drugs has traditionally included drugs targeting ion channels in cardiac myocytes (sodium channel blockers, calcium channel blockers, and QT prolonging drugs, generally potassium channel blockers), β-adrenergic receptor blockers, and a series of drugs with diverse mechanisms used primarily for the therapy of arrhythmias, such as digoxin, amiodarone, magnesium, and adenosine. However, recent studies have demonstrated that other widely used cardiovascular therapies, such as angiotensin-converting enzyme and hydroxy-methyl-glutaryl (HMG) Co-A reductase inhibitors, can also have important antiarrhythmic effects. These effects can include reduction of SCD, an arrhythmic event that represents the final common pathway for many potential disease pathways,17 and prevention of AF. These studies provide novel potential therapies and implicate new signaling pathways in the pathogenesis of arrhythmias and therefore as potential targets for the development of effective antiarrhythmic interventions. One important mechanism underlying variability in response to antiarrhythmic drugs is variability in pharmacokinetics, the result of the processes of drug absorption, distribution, metabolism, and elimination. Variability in pharmacokinetics contributes substantially to variability in efficacy or toxicity when there is a narrow therapeutic window (the margins between doses and plasma concentrations that are associated with efficacy and those associated with toxicity) and when the drug is metabolized or excreted by a single pathway; these criteria apply to many antiarrhythmic drugs. Sotalol and dofetilide are examples of drugs with narrow therapeutic windows and for which the risk of toxicity, namely torsades de pointes, increases with increasing doses. In addition, both drugs are largely excreted by a single pathway: renal excretion. As a result, drug doses need to be adjusted in renal failure to avoid increased risk of torsades de pointes. By contrast, the toxicity of flecainide is also concentration-dependent, but the drug is eliminated by hepatic metabolism and renal excretion. As a result, impaired metabolism or renal dysfunction alone do not generally alter response to the drug, although rare cases of flecainide toxicity owing to inhibited drug metabolism in patients with renal dysfunction have been reported. Table 111-1 identifies drugs that have narrow therapeutic windows and are eliminated by a single pathway, in addition to circumstances under which the meeting of these two clinical conditions can result in serious and often unexpected drug toxicity. N-acetyltransferase (NAT) activity is responsible for the elimination of procainamide. There are two NAT genes: NAT1 and NAT2. NAT1 is expressed in all individuals, but loss of functional alleles has been reported in NAT2. As a result, patients can be divided into rapid and slow acetylators. Slow acetylators have a higher incidence of procainamide-induced lupus syndrome during chronic therapy.18 In addition to metabolizing enzymes, transport across biological membranes is another crucial determinant of drug disposition that has received increasing attention over the last few years. The multi–substrate efflux carrier P-glycoprotein is the most well-studied to date in terms of cardiovascular drugs. P-glycoprotein is the product of the multi-drug resistance 1 (MDR1; ABCB1) gene, and is a member of the ABCB subfamily of ABC (ATP-binding cassette) transmembrane proteins, which have been implicated with drug resistance in cancer. P-glycoprotein is expressed in drug-resistant cancer cells and in many organs such as the gut and kidney, where it has an important role in distribution and elimination. It acts as an efflux pump and in the gut, preventing the entry of toxic compounds, whereas in the liver and kidney it serves to remove xenobiotics from the circulation. Although P-glycoprotein substrates are diverse, there is considerable overlap between the substrates that are transported by P-glycoprotein and those metabolized by CYP3A4/5. One important role for P-glycoprotein in cardiovascular medicine is that it transports cardiac glycosides. A synonymous single-nucleotide polymorphism in exon 26 (C3435T) in the MDR1 gene has been associated with expression of the transporter and variable digoxin concentration.19 Furthermore, correlation of the MDR1 genotype and digoxin uptake in vivo has been described.20 Many drugs inhibit P-glycoprotein activity, and their use with digoxin can lead to increased digoxin plasma concentrations and toxicity; amiodarone, quinidine, verapamil, itraconazole, cyclosporine, and erythromycin are examples.21 One universal principle of pharmacologic therapy is that the best treatment is that targeted specifically to disease mechanisms. As the understanding of the molecular and cellular basis of arrhythmias has evolved, so too has the list of arrhythmias for which specific mechanisms have been defined, and therefore specific antiarrhythmic drug therapies may be indicated (Table 111-2). However, it is the recent advances in the understanding of molecular mechanisms in specific, rare, inherited monogenic arrhythmia syndromes that has provided insight into common arrhythmias, which might also prompt consideration of specific mechanism-based therapies. Table 111-2 A Mechanism-Based Approach to Antiarrhythmic Drug Therapy The inherited arrhythmia syndrome catecholaminergic polymorphic ventricular tachycardia (CPVT) provides a good contemporary example of how a mechanism-based approach for drug therapy can be used. This syndrome is characterized by VT induced by adrenergic stress in the absence of structural heart disease and a high incidence of SCD. Mutations in two causative genes—RYR2, encoding the cardiac ryanodine receptor (RyR2) Ca2+ release channel, and CASQ2, encoding cardiac calsequestrin—have been associated with CPVT.22 Mutations in these two genes destabilize the RyR2 Ca2+ release channel complex in sarcoplasmic reticulum and result in spontaneous Ca2+ release through RyR2 channels leading to delayed afterdepolarizations (DADs), triggered activity, and bidirectional/polymorphic VT.23 Although ICDs are recommended for the prevention of SCD in patients with CPVT,24 painful shocks can trigger further adrenergic stress and arrhythmias; deaths have occurred despite appropriate ICD shocks. Treatment with β-adrenergic blockers has been shown to reduce arrhythmia burden and prevent SCD, but it is not completely effective.25 However, because Ca2+ leakage through ryanodine channel is a common mechanism of CPVT, ryanodine channel block can have a therapeutic effect. This effect led Watanabe et al.26 to assess whether direct inhibition of RyR2 channels by flecainide could be used to treat CPVT. They discovered that flecainide directly inhibits RyR2 channels and suppresses DADs and triggered activity in Casq2 null cardiomyocytes.26,27 Sodium channel block by flecainide can also be effective in preventing CPVT. Flecainide prevents spontaneous VT and inducible VT by exercise or isoproterenol in vivo. Based on these experimental findings, the effects of flecainide in two patients carrying a CPVT-linked mutation in either RYR2 or CASQ2 were examined, and it was found that flecainide suppressed exercise-induced arrhythmias and recurrences of VT/VF.28 Proarrhythmia represents an extreme example of the phenomenon that drug effects vary widely among individuals. The recognition that drugs designed to suppress arrhythmias can, in some patients, actually increase arrhythmias or provoke new ones is probably the most important factor governing the selection and use of antiarrhythmic drugs. Insight into mechanisms leading to proarrhythmia has had important implications for understanding arrhythmogenesis, rational use of antiarrhythmic therapies, selection of patients for specific therapies, and drug development. Furthermore, because proarrhythmia often seems to develop in the absence of clear risk factors, a role for genetics in predisposing to this adverse drug reaction is increasingly being appreciated.29 Proarrhythmia is defined as the generation of new or worsened arrhythmias with drug therapy, but it might not be readily apparent that a drug is, in fact, responsible for an arrhythmia exacerbation. This is a particular problem in patients with advanced heart failure and in whom the spontaneous development of frequent, serious arrhythmias is common even in the absence of drugs. The following sections review three well-recognized proarrhythmia syndromes: digoxin toxicity, drug-induced torsades de pointes, and toxicity related to sodium channel block. These syndromes serve to illustrate gene-drug interactions potentially mediating proarrhythmia risk (Table 111-3). Although it is said that intoxication with digitalis glycosides can produce virtually any arrhythmia, certain arrhythmias should raise suspicion. These include ectopic rhythms such as atrial tachycardia or premature ventricular contractions (PVCs), thought to be due to DAD-related triggered automaticity with sinus slowing or AV nodal block because of the drug’s indirect vagomimetic effects. The major mechanism underlying digoxin toxicity is pharmacokinetic and is related to inhibition of P-glycoprotein activity, a multisubstrate drug efflux carrier (MDR1), by drugs such as amiodarone, quinidine, verapamil, and the azole antifungal agents.30 The molecular target of digoxin is the sodium-potassium ATPase pump, and downstream physiologies that are modulated by pump inhibition include the sodium-calcium exchanger and other signaling pathways involved in intracellular calcium homeostasis. Therefore, candidate genes in which variants can modulate digoxin effects include those encoding sodium-potassium ATPase, the sodium-calcium exchanger, and ones regulating intracellular calcium homeostasis. Accordingly, it is logical to postulate that congenital arrhythmia syndromes that alter intracellular calcium control, such as CPVT31,32 or the ankyrin-B related form of the congenital LQTS,33 predispose to digoxin toxicity. For asymptomatic arrhythmias owing to digitalis toxicity, discontinuation of the drug and observation are probably sufficient. For more advanced cases, the therapy of choice is antidigoxin antibodies. Temporary pacing may be required on rare occasions. The examples of high-risk pharmacokinetics described in this chapter have in common drugs whose elimination depends on a single pathway: propafenone, sotalol, dofetilide. In each case, risk is conferred by absence of redundancy of drug-elimination pathways for that specific drug. The example of drug-induced torsades de pointes is similar; cardiac repolarization is ordinarily a highly redundant process and can be accomplished by IKs, IKr, and probably other mechanisms. Subjects who exhibit marked QT prolongation with IKr-blocking drugs must therefore represent a subset in whom repolarization is highly IKr dependent and thus have lost redundancy in repolarization mechanisms (Figures 111-1, 111-2). This phenomenon has been referred to as “reduced repolarization reserve”16,34 (Figure 111-3) and suggests that multiple often redundant mechanisms maintain normal repolarization, so that minor alterations in function might not be obvious at baseline. For example, a minor reduction in repolarizing current (e.g., owing to a genetic lesion resulting in a small reduction in IKr) might be without consequence because other mechanisms help to maintain a near-normal QT interval; however, such a reduced reserve can become obvious when further stressors to repolarization are superimposed, such as drug challenge, bradycardia, or hypokalemia. Figure 111-1 Continuous recording from a patient who recently started receiving sotalol. During atrial fibrillation, there is irregularity of the ventricular response creating frequent short-long-short cycles, but there is minimal change in QT intervals (top panel). After DC-cardioversion, the QT interval has increased dramatically to 0.64 s (middle panel), and an episode of torsades de pointes is triggered (bottom panel). (Reprinted with permission from Darbar D, Kimbrough J, Jawaid A, et al: Persistent atrial fibrillation is associated with reduced risk of Torsades de Pointes in patients with drug-induced long QT syndrome. J Am Coll Cardiol 51:836–842, 2008.) Figure 111-2 Mechanisms of proarrhythmia associated with HERG blockade. Blockade of the HERG channel produces prolongation of the QT interval (blue) and generates an early afterdepolarization (red) in the cardiac action potential. These changes, which are heterogeneous across the ventricular wall, create a substrate for torsades de pointes. In this example, torsades de pointes degenerates into ventricular fibrillation. (Adapted with permission from Roden DM, Viswanathan PC: J Clin Invest 115(8):2025–2032, 2005.) Figure 111-3 The concept of reduced repolarization reserve. Patient 1 is prescribed a QT-prolonging drug and has minimal or no prolongation of the QT interval. In contrast, patient 2 was given the same QT-prolonging drug at the same dosage and developed marked prolongation of the QT interval; this has been termed reduced repolarization reserve and has been discussed further in the text. Repolarization is a physiologically redundant process such that IKr block will not result in markedly prolonged repolarization—that is, the system displays some “reserve.” However, otherwise subclinical lesions in other components of the repolarization system, such as reduction of IKs owing to genetic factors, hypokalemia can become apparent as marked QT prolongation when IKr is reduced. (Reprinted with permission from Kannankeril et al., 2010.) Other risk factors for torsades de pointes appear pharmacodynamic in nature: female sex, advanced heart disease or left ventricular hypertrophy, bradyarrhythmias, hypokalemia, and recent conversion from AF.16 Each of these can be interpreted in the context of reduced repolarization reserve, and underlying molecular mechanisms have been proposed in some cases (Table 111-4). Thus, hypokalemia can reduce IKr
Standard Antiarrhythmic Drugs
Principles of Antiarrhythmic Therapy
Historical Perspective on Current Antiarrhythmic Drugs and Their Limitations
Classification of Antiarrhythmic Drugs
Pharmacokinetic Principles
Cytochrome P450s and Other Drug Elimination Molecules
Mechanism-Based Approach to Antiarrhythmic Drugs
Arrhythmia
Mechanism-Based Therapy
Mechanism Targeted
AV nodal reentry
AV reentry
Adenosine
Verapamil
Macroreentry using the AV node
Outflow tract VT
β-Blockers
Verapamil, diltiazem
Adrenergic stimulation
Fascicular VT
β-Blockers
Verapamil, diltiazem
Reentry within the His-Purkinje system
Torsades de pointes due to drugs
Pacing
Isoproterenol
K+ supplementation to 4.5-5 mEq/L
IKr block leading to bradycardia-dependent EADs and unstable intraventricular reentry owing to heterogeneity of repolarization
Congenital long QT syndrome, type 1
β-Blockers
Failure of the adrenergically activated K+ current IKs to maintain action potentials short during adrenergic stimulation
Congenital long QT syndrome, type 2
K+ supplementation to 4.5-5 mEq/L
Abnormal action potential prolongation and heterogeneity owing to loss of IKr function
Congenital long QT syndrome, type 3
Sodium channel block: mexiletine, flecainide
Abnormal action potential prolongation and heterogeneity owing to increased inward sodium current during the action potential plateau
Brugada syndrome
ITO block: quinidine, sotalol
Increased heterogeneity of action repolarization owing to loss of sodium channel function and maintained ITO
AF, VF, or T wave oversensing in short QT syndrome
Quinidine, sotalol
Increased outward current, leading to shortened action potentials, owing to abnormally increased IKs, IKr, or IK1
Catecholaminergic polymorphic VT
Flecainide
Directly inhibits RyR2 channels and suppresses DADs and triggered activity; prevents CPVT
Proarrhythmia Syndromes
Digoxin Toxicity
Drug-Induced Torsades de Pointes
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Standard Antiarrhythmic Drugs