Chapter 33 Soft Tissue Sarcomas
Soft tissue sarcomas are rare and unusual neoplasms, accounting for approximately 1% of adult human cancers and 15% of pediatric malignancies. Sarcomas affect more than 10,600 patients/year in the United States and approximately 4000 patients will die each year from inoperable forms of soft tissue sarcoma.1 Sarcomas continue to carry biologic and clinical interest and significance disproportionate to their clinical frequency because of their often clearly defined molecular genetics and the vast expansion of cytogenetic and molecular genetic information that has been discovered over the past 10 years. Soft tissue sarcomas may develop in any anatomic site and this feature, together with the more than 50 histologic types and subtypes, continues to pose a challenge in diagnosis and management. Our understanding of the histology-specific natural history and of the most important clinicopathologic factors that predict outcome and response to therapy has improved our ability to select an optimal treatment plan for the patient diagnosed with soft tissue sarcoma. Most sarcoma types remain resistant to conventional chemotherapy, which means that approximately 40% of newly diagnosed patients will eventually die of disease. Therefore, there exists an urgent unmet need for new therapies targeting the underlying genetic aberrations driving sarcomagenesis. This chapter focuses on the biology, epidemiology, molecular genetics, histopathology, clinical features, and management of soft tissue sarcomas among adults (older than 16 years).
Distribution, Age, and Causes
Most cases of soft tissue sarcoma are thought to be sporadic and their cause is unknown. In rare cases, genetic factors, environmental factors, prior radiation therapy, viral infections, and immunodeficiency have been associated with the development of sarcoma. In addition, sarcomas have been reported to arise in scar tissue, fracture sites, or anatomic regions associated with prior soft tissue trauma. Genetic syndromes such as neurofibromatosis, familial adenomatous polyposis, and the Li-Fraumeni syndrome have all been shown to be associated with the development of soft tissue sarcoma. Desmoid tumors occur in patients with familial adenomatous polyposis, a disorder caused by the germline adenomatous polyposis coli (APC) gene mutation. Malignant peripheral nerve sheath tumors develop in a benign nerve sheath in approximately 2% of patients with neurofibromatosis. The Li-Fraumeni syndrome is a rare, highly penetrant, familial cancer phenotype usually associated with germline mutations in the p53 tumor suppressor gene.2 Of patients with this syndrome, 80% develop cancer by age 45 and soft tissue or bone sarcomas of diverse histology arise as the index tumor in 36% of patients. Heritable retinoblastoma gene (RB1) mutations are associated with an increased risk of bone and soft tissue sarcoma. For example, patients with RB1 mutations have a 36% cumulative incidence over 50 years of developing sarcoma in previously irradiated tissue.3 Most of the excess cancer risks in hereditary retinoblastoma survivors can be prevented by limiting exposures to DNA damaging agents such as radiotherapy, tobacco, and ultraviolet (UV) light,4 emphasizing the importance of avoiding the use of radiation in sarcoma patients with a known germline mutation in RB1.
A soft tissue sarcoma (STS) is one of the most common types of radiation-associated tumors in the general population.5 Although a clear dose-response relationship for radiation-associated malignancies is not established, it is generally accepted that carcinomas arise in tissues exposed to lower doses, whereas sarcomas are induced in heavily radiated tissues (most patients have received 50 Gy or more) in or close to the radiation fields. The median interval between radiation exposure and the development of sarcoma is 10 years (range, 1.3 to 74 years) and this varies by histologic type, with the shortest latency observed in liposarcoma (median, 4.3 years; range, 3 to 17 years), and the longest in leiomyosarcoma (median, 23.5 years; range, 7.0 to 74.0 years).6 Radiation-associated sarcomas are associated with inferior survival compared with sporadic soft tissue sarcoma, even if one adjusts for known prognostic factors such as histologic type, size, age, margin status, and site.6
Several studies have reported an increased incidence of soft tissue sarcoma after relatively high level occupational exposure to phenoxyacetic herbicides, chlorophenols, and dioxins. However, more recent case-control studies have not confirmed this association and have shown no positive correlation between dioxin concentrations and soft tissue sarcoma risk. In fact, sarcoma risk was highest among those having the lowest dioxin level.7,8
Molecular Genetics
Knowledge of the genomic alterations in soft tissue sarcoma is limited to only the most recurrent alterations. These alterations segregate sarcoma into two major groups. The first group consists of sarcoma types with near-diploid, simple karyotypes that bear few chromosomal rearrangements but have pathognomonic alterations, such as the translocations in myxoid round cell liposarcoma (MRC) [t(12;16)(q13;p11), t(12;22)(q13;q12)] and synovial sarcomas (SSs) [t(X;18)(p11;q11)], APC/β-catenin mutations in desmoid tumors, and activating mutations in KIT or PDGFRA in gastrointestinal stromal tumors (GISTs).9–11 Discovery of these last mutations has led to the clinical deployment of imatinib for the treatment of GIST,12 providing a model for genotype-directed therapies in molecularly defined sarcoma subtypes. Conversely, the second group consists of sarcomas with complex karyotypes, including dedifferentiated and pleomorphic liposarcomas, leiomyosarcomas, pleomorphic malignant fibrous histiocytoma, and myxofibrosarcomas. Sarcomas in this group have no known characteristic mutations or fusion genes, although abnormalities are frequently observed in the Rb, p53, and specific growth factor signaling pathways. Thus, a significant subset of soft tissue sarcoma is characterized by recurrent and specific chromosomal aberrations that can be diagnostically and, occasionally, prognostically useful (Table 33-1). The fusion gene translocations include 13 different gene fusions involving the EWS gene or EWS family members (TLS, TAF2N) found in five different sarcomas and 11 other types of fusions in eight other sarcoma types.
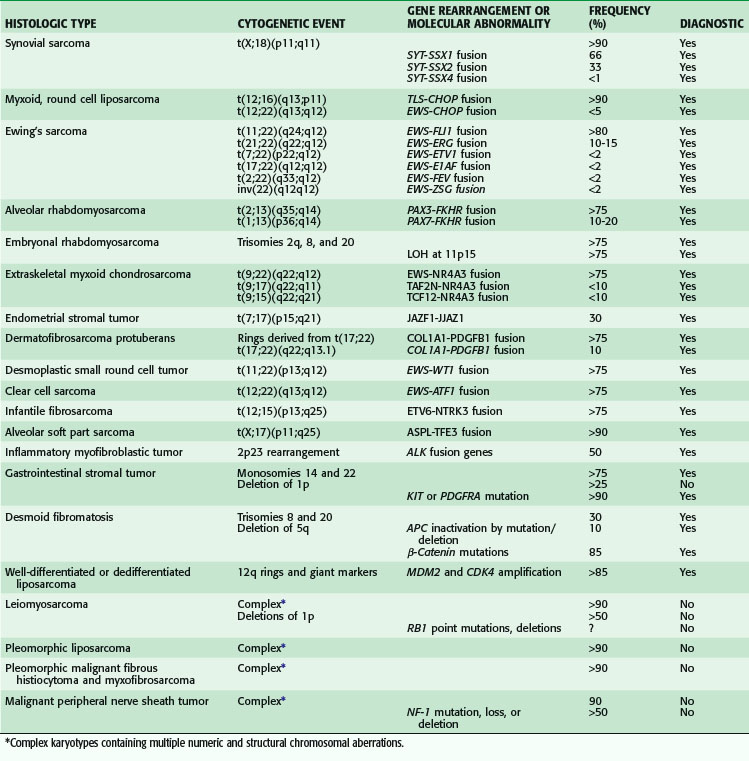
If conventional cytogenetics is not available, molecular genetic techniques (e.g., reverse transcription polymerase chain reaction and fluorescence in situ hybridization) are useful as diagnostic adjuncts. In addition, investigation of molecular changes of genes at the sites of chromosomal alterations has led to the identification of novel genes and the characterization of their mechanisms of deregulation. The tumor suppressor genes best studied in sarcoma are p53, RB1, and NF1. Inactivation of these genes is involved in the tumorigenesis of several sarcomas. The major mechanisms of p53 pathway inactivation in sarcomas include p53 point mutations, homozygous deletion of CDKN2A, which encodes both p14ARF and p16, and MDM2 amplification. In sarcomas with specific reciprocal translocations, p53 pathway alteration is a rare event but, when present, is a strong prognostic factor, associated with significantly decreased survival in synovial sarcoma,13 myxoid liposarcoma,14 and Ewing’s sarcoma–peripheral neuroectodermal tumor (PNET).15 Decreased survival and poor response to chemotherapy in Ewing’s sarcoma–PNET was associated with deletion of CDKN2A, representing a type of p53 pathway alteration through loss of the CDKN2A alternative product p14ARF.15 In contrast, in sarcomas with nonspecific genetic alterations and complex karyotypes, p53 pathway alteration is more common and has weaker prognostic value, often requiring large numbers of patients to achieve statistical significance, as demonstrated in several studies of mixed adult soft tissue sarcoma. Its high prevalence in this class of sarcomas may account for its limited ability to define distinct clinical prognostic subsets in these tumors.
In addition to serving as specific and powerful diagnostic markers, fusion genes resulting from translocations encode chimeric proteins that are important determinants of tumor biology, acting as abnormal transcription factors that alter the transcription of a number of downstream genes and pathways. The structures of these chimeric proteins play a prominent role in the pathogenesis of sarcoma; this has been shown by the impact of relatively minor cytogenetic variability, as a result of variant molecular breakpoints, on tumor phenotype and clinical behavior.16,17 Although the diagnostic significance of sarcoma genomic aberrations has been known for more than 20 years, these same aberrations have only recently become useful as potential therapeutic targets. For example, the identification of the COL1A1-PDGFB1 gene fusion leading to constitutive expression of the platelet-derived growth factor (and its receptor, presumably through an autocrine or paracrine loop) in dermatofibrosarcoma protuberans (DFSP) has paved the way for targeted therapy with imatinib in patients with advanced disease.18,19 The recent discovery that angiosarcomas show distinct upregulation of vascular-specific receptor tyrosine kinases, including TIE1, KDR, SNRK, TEK, and FLT1, by expression profiling and that 10% of patients harbor KDR mutations with evidence for ligand-dependent kinase activation provides a basis for treating angiosarcoma patients with vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitors.20 In a multicenter phase II trial of sorafenib, a small-molecule B-raf and VEGFR inhibitor, in a cohort of patients with metastatic or recurrent sarcoma, only patients with angiosarcoma showed a significant response to therapy, with 5 of 37 patients (14%) having a partial response.21
Evaluation
Evaluation of Extent of Disease
Once the diagnosis and grade are known, evaluation for sites of potential metastasis can be performed. Lymph node metastases occur in less than 3% of adult soft tissue sarcomas. For extremity lesions, the lung is the principal site for metastasis; for visceral lesions and some histologic types of retroperitoneal sarcoma, the liver is the principal site. Thus, patients with low-grade extremity lesions require a chest radiograph and most of those with high-grade lesions require a chest CT scan. CT is the most commonly used modality to evaluate pulmonary metastases. However, it is more expensive than radiographs, delivers a higher radiation dose, and may give false-positive results because of small, indeterminate pulmonary nodules. One study has correlated thoracotomy with CT and found that only 60% of nodules smaller than 6 mm were malignant.22 It is unclear whether there is a better imaging modality to evaluate metastases smaller than 1 cm. Patients with visceral and retroperitoneal lesions should have their liver imaged as part of the initial abdominal CT or MRI. Newer techniques, such as 18F-fluorodeoxyglucose positron emission tomography (FDG-PET), are being used to evaluate distant metastases and, when combined with CT and conventional imaging, may improve the diagnostic accuracy of preoperative staging. However, overstaging remains a problem in 12% of patients and PET-CT remains limited for evaluating pulmonary metastases smaller than 1 cm.23 FDG-PET lacks specificity in its ability to help distinguish between low-grade malignancies and benign entities. An additional concern is that several high-grade sarcoma types, such as round cell liposarcoma and many low-grade sarcomas, do not reliably show uptake for FDG, further limiting its routine use for staging sarcoma patients.
Pathologic Evaluation
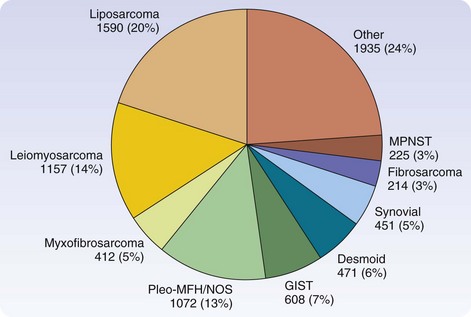
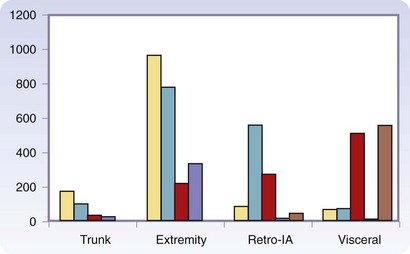
There are more than 50 histologic types of soft tissue sarcoma, many of which are associated with distinctive clinical, therapeutic, or prognostic features. Detailed descriptions of the histopathologic classification and guidelines for the histologic reporting of soft tissue sarcoma have been published elsewhere.24 To summarize, the most commonly found are liposarcoma, leiomyosarcoma, pleomorphic malignant fibrous histiocytoma (pMFH), GIST, desmoids, myxofibrosarcoma, and synovial sarcoma (Fig. 33-1). Histopathology is anatomic site–dependent. The common subtypes in the extremities are liposarcoma, pMFH, myxofibrosarcoma, and synovial sarcoma; in retroperitoneal and intra-abdominal sites, liposarcoma and leiomyosarcoma are the most common histiotypes, whereas in the visceral location, gastrointestinal stromal tumors, leiomyosarcoma, and desmoids are found almost exclusively (Fig. 33-2). Liposarcoma is further classified into three biologic groups encompassing five histologic subtypes, based on strict morphologic features and cytogenetic aberrations—well-differentiated, dedifferentiated, myxoid, round cell, and pleomorphic.24 The well-differentiated and dedifferentiated subtypes account for 42% and 21% of liposarcomas, respectively, and are more commonly found in the retroperitoneal location; the myxoid, round cell, and pleomorphic subtypes account for 25% and 8% of liposarcomas, respectively, and are usually located in the extremity (Fig. 33-3).
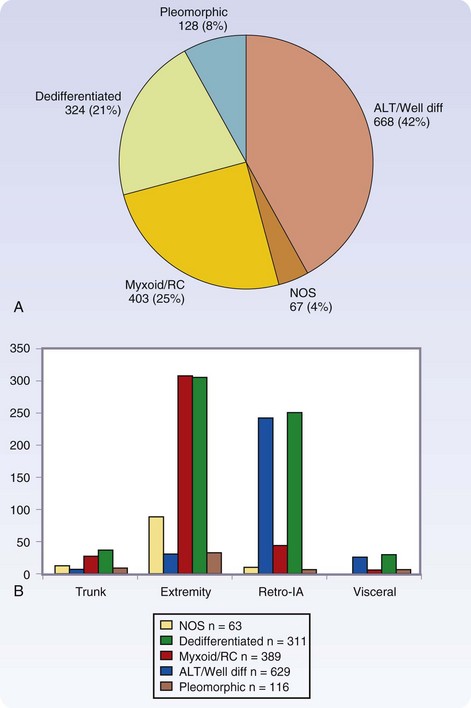
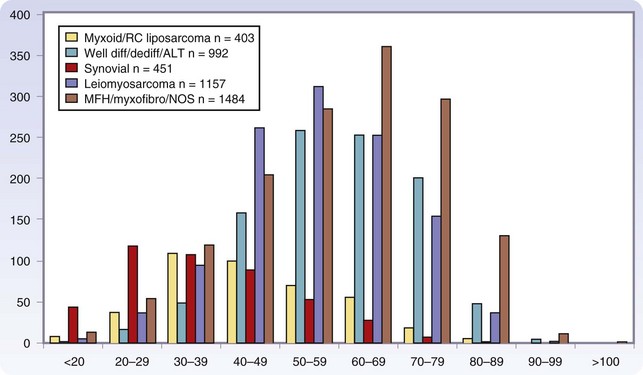
Age is also a factor in histopathology, with the translocation-associated sarcomas often presenting at an age approximately 2 decades earlier than the more complex sarcoma types. In childhood, embryonal rhabdomyosarcoma is most common, synovial sarcoma has a peak incidence among individuals in their 20s and 30s, myxoid and round cell liposarcomas have a peak incidence among those in their 30s, and well-differentiated or dedifferentiated liposarcoma, leiomyosarcoma, pMFH, and myxofibrosarcoma are the predominant types in the older population, with a peak incidence among those in their 50s to 60s (Fig. 33-4). The designation MFH is currently being reevaluated, with many of these tumors being reclassified as myofibrosarcoma, pleomorphic sarcoma, or dedifferentiated liposarcoma.
Because none of the existing grading systems are ideal and applicable to all tumor types, sarcoma histologic type and liposarcoma subtype are generally important determinants of prognosis and predictors of distinctive patterns of behavior. Biologic behavior is currently best predicted based on histologic type, histologic grade, tumor size, and depth. Although many published series have combined all the histologic types of sarcoma, the significance of such subtyping is exemplified by liposarcoma, in which the five subsets (well-differentiated, dedifferentiated, myxoid, round cell, and pleomorphic) have totally different biology, patterns of behavior, and response to chemotherapy.24–27 A further clear demonstration is the importance of myogenic differentiation in pleomorphic sarcomas, which is associated with a substantially increased risk of metastasis.28 In a postoperative nomogram based on a database of 2136 adult patients from Memorial Sloan-Kettering Cancer Center (MSKCC), histologic type was found to be one of the most important predictors of sarcoma-specific death, with malignant peripheral nerve sheath tumors having the highest risk of mortality.29 A more recent liposarcoma-based nomogram has further highlighted the importance of histologic subtype in enhancing assessment of disease-specific survival for the individual patient.30
Histologic Grading and Prognostic Factors for Outcome
The histologic type of sarcoma does not always provide sufficient information for predicting the clinical outcome and, therefore, may be inadequate to inform therapeutic decisions completely. Grading, based on histologic parameters only, evaluates the degree of malignancy and probability of distant metastasis. Several grading systems based on various histologic parameters have been published and proved to correlate with prognosis. The two most important parameters appear to be mitotic index and the extent of tumor necrosis. In general, the two most widely used grading systems are the National Cancer Institute (NCI) system developed by Costa and colleagues and the FNCLCC system developed by the French Federation of Cancer Centers Sarcoma Group. Both are three-tier systems (high, intermediate, and low). A comparative study of these three-tier systems has shown a slightly increased ability of the FNCLCC system to predict distant metastasis development and tumor mortality compared with the NCI system.31 However, studies evaluating the interobserver reproducibility of the FNCLCC system have shown a 60% to 75% agreement for tumor grade and a 61% to 75% agreement for histologic type.32 This high level of disagreement (25% to 40%), even among expert sarcoma pathologists, emphasizes the importance of histologic peer review and the need for developing more objective systems for sarcoma grading and classification. In fact, neither the FNCLCC system nor the NCI system has been formally endorsed by the World Health Organization24 or Association of Directors of Anatomic and Surgical Pathology.
MSKCC researchers have developed a nomogram that adds to the prognostic value of size, grade (low versus high), and tumor depth, with the addition of age, site, and histopathology to refine predictions further of probability of sarcoma-specific survival.29 Although there is widespread use of some form of grading system in the diagnosis and management of sarcomas, there is also agreement that currently no grading system performs well for every type of sarcoma. For a number of reasons, certain histologic types of sarcoma do not lend themselves well to grading. To address some of these issues, my group has recently developed histology-specific nomograms for liposarcoma,30 synovial sarcoma,33 and GIST34 that emphasize the importance of assessing clinical and histologic parameters to improve prognostic accuracy for the individual patient. The limitations of the present grading systems emphasize the need to develop histology-specific molecular or genetic biomarkers that can be combined with conventional clinicopathologic variables to improve objective and accurate assessment of sarcoma prognosis for the individual patient. This would enable the treating physician to design a treatment strategy tailored to an individual patient’s risk of relapse and potential for an aggressive clinical course.



