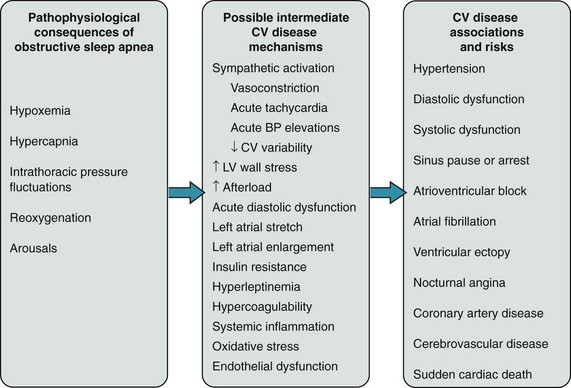
110 Obstructive sleep apnea is characterized by the failure of neuromuscular mechanisms governing upper airway patency during sleep, resulting in airway narrowing or collapse. Although apnea can occur with a person in any position, the upper airway is more vulnerable to obstruction in the supine position because of dorsal displacement of the tongue and soft tissues. Upper airway collapse can occur during any stage of sleep, but susceptibility is greatest during rapid eye movement (REM) sleep. Heralded by atonia of skeletal muscles, including that of the pharynx and upper airway but excluding the respiratory diaphragm and extraocular muscles, REM sleep occurs cyclically (4 to 6 times) throughout the sleep cycle and accounts for about 25% of total sleep time. Of potential pathophysiological importance in arrhythmogenesis, REM is a state of autonomic instability, marked by surges in both sympathetic and vagal activity, with attendant variations in heart rate, blood pressure, and peripheral vascular resistance.1 The best available data from population-based studies that used polysomnographic monitoring in adults provide estimates of the prevalence of OSA of between 3% and 28%.2 Men, in part because of truncal distribution of adipose tissue and muscle, have a higher risk of OSA than women, but the sex prevalence gap narrows after menopause. Age, obesity, and increased neck circumference are additional strong risk factors for OSA.3 Excessive daytime sleepiness, a result of frequent nocturnal central nervous system arousals, is the most important symptom of OSA and a potential contributor to motor vehicle accidents. Yet less than 25% of individuals with OSA actually report excessive sleepiness, although it is likely that subjective reporting under-represents the true symptomatic burden of OSA.4 Daytime sleepiness, even in those with mild OSA, has been shown to correlate with the blood pressure–lowering effects of OSA therapy in patients with systemic hypertension, suggesting an important interaction between cardiovascular disease and OSA that extends beyond the absolute value of the AHI. Other typical signs and symptoms of OSA include loud snoring, interrupted and unrefreshing sleep, and pauses in breathing witnessed by a bed partner. CSA-CSR reflects derangements in ventilatory control mechanisms caused by the hemodynamic consequences of left atrial hypertension and decreased cardiac output (usually caused by left ventricular dysfunction). Partly because of irritant receptor stimulation by pulmonary microvascular congestion, heart failure patients chronically hyperventilate, resulting in low blood carbon dioxide tensions during wakefulness.5 The relative hypocapnia, together with a heightened chemoreflex response to carbon dioxide and prolonged circulation time, destabilizes the ventilatory control system during sleep and results in CSA-CSR. As would be expected by these mechanisms, inhalation of a carbon dioxide–enriched gas abolishes CSA. CSA-CSR, like OSA, is diagnosed by multichannel polysomnography and is quantified by the central AHI. The characteristic waxing and waning pattern of ventilation, which also may be seen during wakefulness, is most prominent during non-REM sleep and is characteristically absent during REM sleep, where cortical influences on ventilation often override the chemoreflex. Similar to OSA, CSA-CSR is associated with repetitive cycles of acute stressors, including hypoxemia, hypercapnia, sympathetic activation, and fluctuations in heart rate and blood pressure.6 In contrast to OSA, the influence in CSA-CSR of these processes on cardiovascular outcomes, including arrhythmias, has been relatively unexplored. Some data suggest that CSA-CSR contributes to worse cardiovascular outcomes, including mortality, above and beyond what is attributable to accompanying heart failure. What is known regarding the effect of CSA-CSR on specific arrhythmia syndromes will be discussed next. The prevalence of CSA-CSR in the general population or in unselected individuals with left ventricular dysfunction is unknown. In two studies of more than 500 patients with predominantly New York Heart Association class II heart failure and an average left ventricular ejection fraction of approximately 25%, the prevalence of sleep apnea identified by polysomnography was between 33% and 40%.7,8 The prevalence of CSA-CSR increases directly with the severity of heart failure. Like OSA, age and male sex increase the risk for CSA-CSR, with narrowing of the gender gap after menopause. Obesity, diabetes, dyslipidemia, hypertension, ischemic heart disease, and systolic and diastolic heart failure all cluster with sleep apnea (Figure 110-1).9 Simply by association with these conditions, the risk of arrhythmias is increased in patients with OSA and CSA. There are multiple pathophysiological mechanisms by which sleep apnea may directly promote the initiation or maintenance of specific clinical arrhythmias (discussed next). Figure 110-1 Obstructive sleep apnea (OSA) and cardiovascular disease mechanisms. The pathophysiology of OSA can acutely and chronically elicit multiple intermediate cardiovascular disease mechanisms, which can promote the association of OSA with a number of cardiovascular conditions and diseases. (Reproduced from Gami AS, Somers VK: Sleep disorders and cardiovascular disease. In Libby P, Bonow RO, Mann DL, et al, editors: Braunwald’s heart disease: a textbook of cardiovascular medicine, ed 8, Philadelphia, 2007, Saunders.) In individuals with OSA, apnea and hypoxemia activate the physiologic “diving reflex,” so named from observations of oxygen conservation during prolonged water submersion by marine mammals. Hypoxemia, via stimulation of the carotid bodies, normally not only induces hyperventilation but also results in bradycardia. With normal breathing and hyperventilation, this slowing of the heart rate is attenuated by the vagolytic effects of lung inflation. This latter buffer is lost during apnea and when coupled with the effects of oxyhemoglobin desaturation, results in marked bradycardias.10 Electrophysiological studies in individuals with OSA and bradycardias referred for permanent pacemaker implantation showed no evidence of significant sinus, atrioventricular node, or His-Purkinje system disease, reflecting the influence of autonomic changes that occur in OSA. Multiple pathophysiological mechanisms may directly link OSA and AF. Increased left atrial size is associated with OSA independently of other medical conditions.11 Otto and colleagues compared left atrial volume indices in individuals with OSA and without OSA who otherwise were completely healthy, middle-aged men and women. They found that the left atrial volume index was normal in all subjects, yet still significantly higher in the individuals with OSA. Left atrial size may be increased in individuals with OSA because of its association with hypertension, for which it has been identified as a secondary cause, and diastolic dysfunction.12 Even in otherwise healthy individuals, subtle changes of diastolic dysfunction are present in those with OSA, and the severity of diastolic dysfunction correlates directly with the severity of oxygen desaturation. Concomitant obesity likely contributes to an enlarged left atrium as a result of a larger body mass and increased total blood volume. Left atrial size may also be increased in OSA because of repetitive inspiratory efforts against an obstructed upper airway, which produce wide fluctuations in intrathoracic pressure that increase cardiac wall stress.13 This process may also cause chronic stretching of the atrium, particularly at anchoring regions such as the pulmonary vein ostia, leading to electrical and structural remodeling. Activation of stretch-sensitive ion channels in the atrium may have implications for the initiation of AF. Another potentially important mechanism of AF is the marked fluctuations in autonomic tone that occurs in OSA. Cessation of breathing is associated with increases in sympathetic neural output as measured by peripheral microneurography.14 Via the diving reflex, there may also be a concomitant rise in cardiac vagal activity.10 Furthermore, surges in sympathetic neural activity occur with each central nervous system arousal terminating an apnea. Although sleep stage–dependent mechanisms have yet to be proven in arrhythmogenesis, the influence of sleep stage on autonomic tone is well-recognized. Stage II sleep is punctuated by characteristic “K-complexes,” high-amplitude electroencephalographic discharges associated with transient increases in peripheral sympathetic neural activity, and a rise in blood pressure.1 Slow-wave sleep, underrepresented in severe OSA, is a state of parasympathetic predominance. REM sleep is associated with abrupt fluctuations in both sympathetic and parasympathetic activity. Although it is clear that the autonomic system plays a major role in the pathophysiology of AF, its specific effects are still being determined.15 The processes that occur in OSA may lead to activation of atrial catecholamine-sensitive ion channels, which could result in focal discharges that initiate AF, and also to vagally mediated changes in atrial conduction properties that promote and maintain AF. In addition, individuals with OSA have chronically elevated sympathetic activity even during the waking period, which may have a deleterious effect on rate-control strategies for AF management.14 OSA is associated with systemic inflammation, evidenced by increases in levels of C-reactive protein, serum amyloid A, and interleukins, which may have implications for atrial fibrosis and remodeling.16 Recently, several measurable electrophysiological abnormalities that promote AF have been demonstrated in patients with OSA. The presence and severity of OSA correlate with interatrial and left atrial electromechanical delay as measured by tissue Doppler imaging and P-wave dispersion.17 Furthermore, atria in patients with OSA demonstrate lower voltage, slower conduction, and more widespread, complex fractionated electrograms.18
Sleep-Disordered Breathing and Arrhythmias
Obstructive Sleep Apnea
Pathophysiology
Epidemiology and Clinical Features
Central Sleep Apnea
Pathophysiology
Epidemiology and Clinical Features
Mechanisms of Arrhythmias in Sleep Apnea
Bradyarrhythmias
Atrial Fibrillation
Sleep-Disordered Breathing and Arrhythmias



