(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Ischemia and postischemic reperfusion cause a wide array of functional and structural alterations of mitochondria. Although mitochondrial impairment is recognized as pivotal in determining loss of cell viability, the causal relationships among the various processes involved is not well defined. Nevertheless, there is a consensus in attributing a crucial role to opening of the mitochondrial permeability transition pore (MPTP). Strong support for this concept has recently been provided by the reduced infarct size in mice lacking cyclophilin D. This protein located within the mitochondrial matrix favors MPTP opening by increasing its sensitivity to Ca2+ in a process that is antagonized by cyclosporin A. Genetic approaches have also been used to demonstrate that adenine nucleotide translocase is not an essential component of the MPTP. Key factors linking mitochondrial dysfunction to myocardial injury have been studied along with potential protective mechanisms elicited by endogenous processes and pharmacological treatments. In particular, a reduced rate of ATP hydrolysis and a slight increase in reactive oxygen species (ROS) formation appear to represent the prevailing components of self-defense mechanisms, especially in the case of ischemic preconditioning. These cardioprotective processes are activated by signaling pathways, which converge on mitochondria activating the mitochondrial KATP channels and/or inhibiting the MPTP. These pathways can also be stimulated by pharmacological treatments. Another major goal for cardioprotection is decreasing the burst in mitochondrial ROS formation associated with postischemic reperfusion. Finally, given an inhibition of fatty acid oxidation associated with cardioprotection, mitochondrial targets for therapeutic intervention may include the switch of substrate being utilized.
Introduction
Acute coronary occlusion as appears in acute coronary syndromes results in ischemia/reperfusion in the heart and is a major cause of mortality in Western societies. Multiple lines of evidence demonstrate that increased oxidative stress and mitochondrial dysfunction are key mediators of the cardiac dysfunction induced by ischemia/reperfusion [1, 2]. Mitochondrial dysfunction can affect cell viability through a variety of pathways. Loss of ATP synthesis and increase of ATP hydrolysis, impairment in ionic homeostasis, formation of reactive oxygen species (ROS), as well as the release of proapoptotic proteins constitute the key factors in the generation of irreversible damage [3–5]. These series of events explain why mitochondria are involved in both necrosis and apoptosis following postischemic reperfusion. Although there is general consensus on the role of mitochondria in cell death, important questions remain unsolved, especially concerning the molecular mechanisms and causal relationships.
In recent years, a great interest has been diverted to the mitochondrial permeability transition pore (MPTP) [6–9]. A role of the MPTP in the reperfusion injury of the heart has been hypothesized at the end of the 1980s [10, 11] and subsequently demonstrated in isolated cardiomyocytes [12] as well as in perfused hearts [13]. MPTP opening has also been proposed to play a role in both ischemic preconditioning (IPC) and postconditioning [14, 15]. Mitochondria likely plays important functional roles in endogenous mechanisms of protection as well [16]. It has been demonstrated that even a slight increase in ROS formation is associated with a boosting of self-defense mechanisms [17]. Indeed, antioxidants abrogate the powerful protection afforded by IPC [17]. It has been proposed that mitochondria are involved in this protective mechanism(s) through the opening of both KATP channels [18] and the MPTP [19].
Cardioprotection activates a number of signaling pathways that reduce the level of triggers (e.g., ROS species and calcium) or enhances inhibitors of the MPTP at the start of reperfusion. Due to their multifaceted relationship with cell death, mitochondria appear to be ideal targets for interventions aimed at preserving cardiomyocyte viability [4]. Besides self-defense mechanisms, pharmacological approaches have been developed to tackle myocardial injury directly at the mitochondrial level. The majority of approaches proposed so far can be grouped into three major cardioprotective strategies, namely, MPTP inhibition, decreased ROS formation, and inhibition of fatty acid oxidation (FAO). These approaches will also be discussed in this chapter.
Mitochondria in Ischemia and Reperfusion in the Heart
Oxygen is the main acceptor of electrons harvested from nutrients, mainly carbohydrates and lipids, through the activity of NAD- and FAD-dependent dehydrogenases. The reduced forms of these coenzymes are reoxidized at the level of complexes I and II of the respiratory chain, and the electrons are funneled to complex IV where more than 90% of the intracellular oxygen is reduced to water. Thus, oxygen is for the most part utilized in a single reaction, and this explains the profound metabolic changes occurring when oxygen supply is no longer sufficient for the activity of cytochrome c oxidase. In fact, the very first and immediate consequence of an insufficient cellular oxygenation is the inhibition of electron flow along the respiratory chain that inevitably impairs both energy conservation and oxidative metabolism (Fig. 16.1).
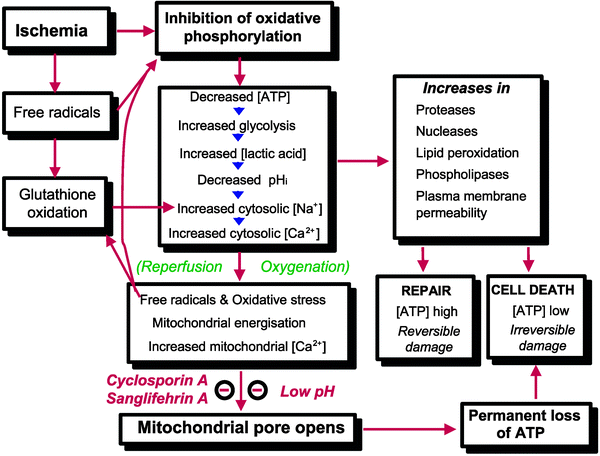
Fig. 16.1
Factors leading to opening of the mitochondrial permeability transition pore in reperfusion. For more details see the text (From Halestrap et al. [7] with permission from Oxford University Press)
Impairment of Energy Metabolism
The energy released from the flux of electrons from reducing to oxidizing components of the respiratory chain is converted into the extrusion of protons from the mitochondrial matrix into the intermembrane space generating a proton motive force (Δp) that consisted of membrane potential (Δψm) and pH gradient (ΔpHm) components. The utilization of this proton motive force allows ATP synthesis, maintenance of ion homeostasis, and protein import for mitochondrial biogenesis. Therefore, by inhibiting electron flow the lack of oxygen prevents ADP phosphorylation into ATP at the level of F1Fo ATP synthase. Importantly, this enzyme catalyzes a reversible process of coupling between proton movements across the inner mitochondrial membrane (MIM) and ATP synthesis or hydrolysis. In fact, if the proton gradient is not generated by the respiratory chain, F1Fo ATP synthase couples ATP hydrolysis with proton pumping to maintain the Δψm. This process perhaps explains why this enzyme is sometimes also termed ATPase, and this reverse operation resembles the activity of other cellular ATPases that convert ATP hydrolysis into active transport of ions against concentration and/or electrical gradients. The net result is that the mitochondria cease to be the main source of intracellular ATP but rather become a powerful system for hydrolyzing glycolytically produced ATP [20, 21]. Under anoxic/ischemic conditions the inhibition of ATP hydrolysis is likely a major component of myocardial protection elicited by both self-defense mechanisms and pharmacological treatments [22].
Impairment of Oxidative Metabolism
The degradative oxidation of nutrients catalyzed by anaerobic dehydrogenases is made possible by the reduction of the oxidized forms of pyridine and flavin nucleotides (i.e., NAD(P)+ and FAD). Once reduced, these coenzymes must be immediately reoxidized, since their intracellular contents are not abundant. In addition, they cannot be replenished by extracellular sources since they cannot permeate the cell membrane, at least at the turnover rates dictated by catabolic pathways. Since the main pathway for NADH(H+) and FADH2 reoxidation is the respiratory chain, its inhibition hampers the oxidation of all substrates. On the other hand, in the presence of respiratory chain inhibition, NADH(H+) oxidation is mostly catalyzed by lactate dehydrogenase. This allows the prosecution of cytosolic anaerobic glycolysis which is practically the only pathway for ATP (re)synthesis under anoxic conditions, especially considering that phosphocreatine is rapidly depleted at the onset of ischemia [23, 24]. The arrest of FAO results in the accumulation of potentially toxic intermediates, such as long-chain acyl-CoAs and long-chain acylcarnitines [25, 26]. In addition, the formation of these metabolites exacerbates metabolic impairments by reducing the availability of unesterified CoASH and carnitine. These primary modifications induce a wide array of changes. Among the latter processes, a prominent role is attributed to impaired Ca2+ homeostasis and increased ROS formation.
Alterations in Mitochondrial Calcium Homeostasis
Since both mitochondrial Ca2+ uptake and release depend on Δp [27], respiratory chain inhibition results in profound alterations of mitochondrial Ca2+ homeostasis that can trigger and be further exacerbated by opening of the MPTP [6–8, 27]. It is generally accepted that during ischemia and/or reperfusion mitochondrial [Ca2+] ([Ca2+]m) increases, yet it must be pointed out that quite different mechanisms underlie [Ca2+]m rise or overload. In fact, during ischemia Δp is abolished because of respiratory chain inhibition, and [Ca2+]m may passively follow elevations of cytosolic Ca2+. On the other hand, upon reperfusion, Δp is recovered along with the capacity of mitochondria to take up huge amounts of Ca2+. Therefore, only under the latter conditions active accumulation of Ca2+ can take place within the matrix, which is more likely to cause MPTP opening, mitochondrial depolarization, impairment of ATP synthesis, and the initiation of massive hydrolysis of ATP. Regardless, [Ca2+]m elevation is the consequence of a preceding rise in cytosolic Ca2+. Therefore, the abrupt release of Ca2+ from an overloaded matrix is not likely to determine per se intracellular Ca2+ overload. The lack of adequate pharmacological or genetic tools to inhibit mitochondrial Ca2+ uptake in situ has so far prevented a clear assessment of its role in myocardial physiology and pathology. Relevant questions, such as mitochondrial involvement in excitation-contraction coupling or the relationships between mitochondrial Ca2+ uptake and cell injury, remain unanswered. Nevertheless, strategies aimed at decreasing cytosolic Ca2+ overload are likely to prevent or delay alterations in mitochondrial structure and function.
Increased Generation of Reactive Oxygen Species
Reactive oxygen species (ROS) are formed within mitochondria under physiological and pathological conditions, especially during postischemic reperfusion [28, 29]. A frequent estimate that 2–4% of the oxygen utilized by the respiratory chain undergoes univalent reduction becoming superoxide anion is likely to be an overestimation. It has been suggested that a correct estimate might be one order of lower magnitude [30, 31]. Superoxide anion formed at the level of complexes I and III is rapidly transformed into hydrogen peroxide (H2O2) by a family of metalloenzymes, superoxide dismutases (SOD) [32]. The inhibition of the respiratory chain favors ROS formation lending support to the concept that respiratory complexes, especially complex I, are the main intracellular sites for ROS formation. However, mitochondrial sites other than the MIM are capable of generating H2O2 at significant rates. A relevant role in this respect is likely played by monoamine oxidases (MAO). These mitochondrial outer membrane (MOM) flavoproteins catalyze electron transfer from various amine compounds (including the catecholamines) to O2, thus producing large amounts of H2O2 [33]. One more example of mitochondrial ROS generation at sites other than the respiratory chain is provided by p66Shc. This protein, which localizes in part within mitochondria, catalyzes electron transfer from cytochrome c to oxygen. The consequent formation of ROS has been linked to MPTP opening and triggering of apoptosis [34]. Fibroblasts lacking p66Shc have displayed a reduced formation of ROS in response to oxidative stress-inducing agents. Conversely, p66Shc overexpression has resulted in enhanced stress-induced apoptosis [34, 35]. Of interest, targeted mutation of the mouse gene encoding p66shc induces stress resistance and prolongs life span [36]. More recent work suggests that protein kinase C beta (PKC β)-mediated phosphorylation of p66Shc on Ser36 could cause its translocation to mitochondria [37]. PKC β, activated by oxidative conditions in the cell, phosphorylates p66Shc; phosphorylated p66Shc is recognized by the prolyl isomerase Pin1 and accumulated in mitochondria. Once imported, p66Shc causes alterations of mitochondrial Ca2+ responses and three-dimensional structure leading to inducing apoptosis. Therefore, the increase in mitochondrial ROS formation caused by p66Shc serves to amplify PKC β signaling triggered by an initial oxidative stress. The above-mentioned alterations trigger and underlie multiple deleterious consequences of ischemia, and their role is likely to be crucial for any myocardial disease. Considering mechanisms involved in cell death, MPTP opening and release of apoptogenic proteins represent the most relevant processes.
Mitochondrial Permeability Transition Pore Opening
The MPTP is a voltage-dependent, high-conductance channel located in the mitochondrial inner membrane (MIM). In the fully open state, the apparent pore diameter is 3 nm, allowing passive diffusion of solutes with molecular masses up to about 1.5 kDa (see also Chap. 3) [27, 38]. A relevant feature of the MPTP is its inhibition by cyclosporin A (CsA). Since the effect of CsA can be relieved by increasing the Ca2+ load [27], the effect of CsA can best be described as “desensitization” of the MPTP to Ca2+. This is a key point because the MPTP can still be opened in the presence of CsA, a fact that mandates careful controls in order to assess whether the MPTP is actually inhibited after administration of CsA in vitro and in vivo [39]. Because their masses are larger than the MPTP exclusion limit, proteins do not diffuse through the pore. Consequently, when mitochondria are suspended in a crystalloid buffer, matrix proteins exert a colloid osmotic pressure that causes mitochondria to swell. Even a large increase in matrix volume does not damage the MIM, which is protected by cristae unfolding. Pore closure in saline media is followed by MIM refolding and full recovery of function, provided that cytochrome c is added back [40]. On the other hand, matrix swelling may cause the rupture of the mitochondrial outer membrane (MOM) and release of proteins from the intermembrane space. An important member of this group is cytochrome c, which after binding Apaf-1 in the cytosol causes the activation of caspase 9, triggering the apoptotic cascade [3, 41]. Importantly, when the matrix volume is normal, the largest fraction of cytochrome c is sequestered within the MIM cristae [42]. Thus, in the absence of matrix expansion, only a small fraction of cytochrome c is available for its release through the MOM pores formed by proapoptotic members of the Bcl-2 family [43]. This notion suggests that MPTP is also relevant for the intramitochondrial redistribution of cytochrome c in cells undergoing apoptosis when the entire pool of this chromoprotein is released from mitochondria.
Mitochondrial swelling is generally considered as a consequence of MPTP opening. However, it must be pointed out that swelling might not occur in intact cells due to the high cytosolic content of proteins and macromolecules [27]. However, short open times and/or lower conductance states of MPTP might not cause detectable changes in matrix volume [44]. Indeed, different MPTP-opening states have been demonstrated in isolated mitochondria and in intact cells. A subconductance state has initially been proposed based on finding showing that mitochondria impermeable to sucrose could still release Ca2+ in a process that occurred in the absence of added Na+ and has not been inhibited by ruthenium red [45]. These findings also indicate that the MPTP is independent of Na+/Ca2+ exchanger and Ca2+ uniporter. Subsequently, electrophysiological measurements performed on isolated mitochondria have demonstrated that, besides the conductance of 1.200 pS of the MPTP in its fully open state, a half conductance of 500 pS is detected [38]. Such a lower conductance would correspond to an exclusion size of <300 Da thereby allowing ion diffusion [44]. It has been proposed that the pore in this conformation should flicker allowing the release of accumulated Ca2+ in a pH-dependent process favored by matrix alkalinization [44]. The efflux of Ca2+ by means of MPTP opening would be advantageous since the consequent depolarization would prevent an immediate reuptake of the released Ca2+. The function of MPTP as a Ca2+ release channel might explain the increase in calcium detected in mitochondria isolated from CsA-treated cardiomyocytes [46]. In addition, the results of elegant experiments performed in isolated cells suggested the involvement of MPTP in the amplification of the Ca2+ signals primarily originating from the endoplasmic reticulum [44, 47].
A key pathologic role in cell death is attributed to MPTP opening as related to a wide range of pathologies extending far beyond myocardial injury [27]. MPTP opening can be induced by almost all of the conditions discussed above, i.e., [Ca2+]m increase, Δψm fall, and oxidative stress (Table 16.1). ROS-induced ROS release might be particularly relevant for the amplification of an initial oxidative stress resulting in the recruitment of a great proportion of mitochondria in an injured cell. In addition, other MIM-localized anion channels might also contribute to the intracellular spreading of ROS-induced mitochondrial depolarization [48]. These processes appear to represent a major cause of mitochondrial dysfunction creating conditions that are not compatible with cell survival. This especially holds true in the case of prolonged MPTP openings [49] that cause collapse of Δψm, depletion of ATP and NAD+, matrix swelling, and rupture of the MOM followed by release of proapoptotic proteins from the intermembrane space, such as cytochrome c.
Table 16.1
Physiologic effectors of MPTP
Factors | Effects |
|---|---|
Divalent cations | The permeability transition is favored by accumulation of Ca2+ in the matrix and counteracted by Me2+ ions [27] |
Inorganic phosphates | Favors MPTP opening |
Δψm | At physiological membrane potentials, the pore favors the closed state, while it can be opened by membrane depolarization [80] |
ROS | |
Adenine nucleotides | Decrease probability of pore opening [114] |
The opening of MPTP is favored by [Ca2+]m elevation, depolarization, and increases in ROS and inorganic phosphate (Pi). These factors are counteracted by physiological MPTP antagonists, such as elevated values of Δψm and high concentrations of H+, Mg2+, and adenine nucleotides, especially ADP [7, 27]. This perhaps explains the observations reported from several laboratories that MPTP opening occurs during postischemic reperfusion. In fact during ischemia, intracellular acidosis along with increase in levels of Mg2+ and ADP can override the MPTP-opening conditions established by Δψm decrease and increases in Ca2+ and Pi levels. Conversely, upon reperfusion the recovery of pH together with a burst in ROS formation in the presence of high-matrix concentrations of Ca2+ and Pi creates the perfect scenario for MPTP opening despite the antagonizing effect of Δψm recovery (Fig. 16.1).
In isolated mitochondria, MPTP opening usually occurs upon additions of Ca2+ exceeding 0.1 mM. Such high Ca2+ concentrations are rarely achieved in viable cells suggesting that Ca2+ does not cause MPTP opening directly, but MPTP is rather sensitized to Ca2+ by several processes, such as formation of ROS or metabolites generated by Ca2+-dependent enzymes. Previous observations have suggested that MPTP onset is due to a deadly mixture of ROS generation and pH normalization, whereas intracellular Ca2+ overload would be the consequence rather than the cause of bioenergetic failure [50]. On the other hand, the activation of Ca2+-dependent enzymes could signal the occurrence of cell damage to mitochondria by converting and amplifying initial Ca2+-related signals into Ca2+-dependent derangements. Among the possible signaling pathways, it has been characterized the activation of cytosolic phospholipase A2 resulting in the formation of arachidonic acid, which promotes MPTP opening, triggering apoptosis [51].
Several reports have indicated the involvement of calpains in these processes. The relationship between these Ca2+-dependent proteinases and cell death involves multiple processes and cellular sites [52, 53]. It has been reported that intramitochondrially localized calpain l isoform [54] may be implicated in MPTP opening [55], mitochondrial release of the apoptosis-inducing factor (AIF) [56], and cleavage of Bid, a proapoptotic member of the Bcl-2 family [57]. Of interest, calpain activation within mitochondria appears to be triggered by interaction with its natural activator, acyl-CoA-binding protein [54]. Therefore, counteracting [Ca2+]i rise and/or activation of Ca2+-dependent enzymes can represent a potentially effective strategy for preventing detrimental MPTP opening.
ATP-Binding Cassette Mitochondrial Erythroid
Recently, mitochondrial transporter ATP-binding cassette mitochondrial erythroid has been found to be a novel gene necessary for cardiac recovery after ischemia/reperfusion [58]. ATP-binding cassette mitochondrial erythroid (ABC-me; ABCB10; mABC2) is a mitochondrial transporter highly induced during erythroid differentiation and predominantly expressed in bone marrow, liver, and heart. Mice harboring one functional allele of ABC-me (ABC-me+/−) have been generated by replacing ABC-me exons 2 and 3 with a neomycin resistance cassette.
Under basal conditions, ABC-me+/− mice have normal heart structure, hemodynamic function, mitochondrial respiration, and oxidative status. After ischemia/reperfusion, the recovery of hemodynamic function has been reduced by 50% in ABC-me+/− hearts as a result of impairments in systolic function. This reduction has been associated with impaired mitochondrial bioenergetic function and with oxidative damage of both mitochondrial lipids and sarcoplasmic reticulum Ca2+-ATPase after reperfusion. Treatment of ABC-me+/− hearts with the superoxide dismutase/catalase mimetic EUK-207 has prevented oxidative damage of mitochondria and sarcoplasmic reticulum Ca2+-ATPase and restored mitochondrial and cardiac function to wild-type levels after reperfusion. Inactivation of one allele of ABC-me therefore enhances the susceptibility to oxidative stress induced by ischemia/reperfusion, leading to increased oxidative damage of mitochondria and sarcoplasmic reticulum Ca2+-ATPase and to impaired functional recovery.
Mitochondria and Cardioprotection
Cardioprotection, such as preconditioning and postconditioning, has been shown to result in a significant reduction in cell death. Many of the signaling pathways activated by cardioprotection have been elucidated, but there is still a lack of understanding of the precise mechanisms by which these signaling pathways can reduce cell death. Mitochondria have been reported to be an important player in both apoptotic and necrotic mode of cell death. If mitochondria play an important role in cell death, then it seems reasonable to consider that cardioprotective mechanisms might act, at least in part, by opposing the mitochondria-mediated cell-death pathways. As discussed in previous sections, one of the major mechanisms of cell death in ischemia-reperfusion has been purported to be the opening of a large conductance pore in the MIM, known as the MPTP. Inhibition of this mitochondrial pore appears to be one of the major mechanisms, by which cardioprotection can reduce cell death. Cardioprotection activates a number of signaling pathways that reduce the level of triggers, such as ROS species and calcium, or enhances inhibitors of the MPTP opening at the initiation of reperfusion [59]. These mechanisms will be discussed in the following sections.
Mitochondria Self-Defense Mechanisms
Due to their multifaceted relationship with cell death, mitochondria appear to be ideal targets for interventions aimed at preserving cardiomyocyte viability. Obviously, in the absence of oxygen, mitochondrial ATP synthesis cannot be restored or increased; therefore, protection can result from decreasing ATP hydrolysis. This goal can be achieved through several mechanisms that are also a part of self-defense mechanisms triggered by ischemic preconditioning (IPC). For instance, mitochondrial depolarization promotes F1Fo ATPase binding to its natural inhibitor ATPase inhibitory factor 1 (IF1). This direct inhibition has been proposed to play an essential role in both classical and pharmacological IPC [60]. Mitochondrial ATP hydrolysis appears to be modulated by Bcl-2 since Bcl-2 overexpression in mouse hearts has been found to increase cardiomyocyte survival upon reperfusion and reduce the decline in ATP during ischemia. This effect has been attributed to a direct inhibition of F1F0 ATPase, since the addition of oligomycin did not cause any additional effect [61]. This finding extends the role of Bcl-2 to modulation of mitochondrial metabolism [22]. Interestingly, Bcl-2 is upregulated in preconditioned hearts, while it is downregulated by ischemia/reperfusion [62]. In addition, its downregulation using antisense oligonucleotides has abolished IPC cardioprotection in perfused rat hearts [63].
ATP hydrolysis could also be prevented by MPTP inhibition, a beneficial effect that would add the ATP sparing effect to a wider range of protective actions, such as prevention of NAD+ depletion, preservation of Ca2+ homeostasis, as well as a relative lack of release of proapoptotic proteins [7, 8, 64]. The probability of MPTP opening is reduced in preconditioned hearts upon postischemic reperfusion, which confers a stress-tolerant state to cardiomyocytes [14, 63]. The activation of signaling pathways appears to contribute to reducing the probability of MPTP opening in preconditioned cardiomyocytes [65].
In addition to the protection afforded by MPTP inhibition, numerous studies attribute a protective role to opening of the mitochondrial KATP channel (mitoKATP) [18, 66]. Upon mitoKATP opening the electrophoretic influx of K+ into the matrix causes mitochondrial depolarization along with matrix swelling and alkalinization [67]. It has been suggested that the matrix contraction caused by the Δψm decrease occurring during anoxia would be compensated by the matrix swelling resulting from mitoKATP opening. This type of swelling might both increase the efficiency of energy metabolism in the normoxic myocyte and accelerate the recovery of optimal ATP concentrations during postischemic reperfusion by preventing the loss of substrate channeling otherwise caused by matrix contraction and expansion of the intermembrane space [68]. Since cardioprotection is elicited by both MPTP inhibition and mitoKATP opening, these processes might be interconnected. Mitochondrial depolarization and ROS formation have been suggested as plausible links rendering mitoKATP an additional mode for modulating the MPTP. The decrease in mitochondrial membrane potential caused by mitoKATP opening could reduce the driving force for mitochondrial Ca2+ uptake, thus decreasing the probability of MPTP opening [69]. On the other hand, matrix alkalinization has been suggested to be responsible for the increase in H2O2 formation observed in cardiomyocytes treated with the mitoKATP opener diazoxide [17, 70]. The increased formation of ROS might have reduced the probability of MPTP opening by affecting the redox state of critical thiol groups [71]. In addition, a recent report suggests that PKCε activates mitoKATP resulting in a slight elevation of ROS, which eventually causes MPTP inhibition [72, 73]. The ROS-mediated link between PKCε and mitoKATP might involve additional factors. For instance, the increase in ROS formation induced by diazoxide is blunted in cardiomyocytes and hearts of heterozygous connexin 43-deficient (CX43+/−) mice [74]. This protein forming gap junctions within the sarcolemma has been reported to accumulate within the MIM as a result of IPC [75]; accordingly, IPC protection is lost in CX43+/− mice [76].
Mitochondria-Directed Cardioprotection Strategies
Besides self-defense mechanisms, pharmacological approaches have been developed to tackle myocardial injury directly at the mitochondrial level. The majority of approaches proposed so far can be grouped into several broad cardioprotective strategies including MPTP inhibition, decreased ROS formation, and inhibition of FAO.
MPTP Inhibition
Opening of the MPTP is inhibited by cyclosporin (Cs) A after binding to cyclophilin (CyP)-D, a matrix peptidyl-prolyl cis-trans isomerase [77]. The very high-affinity binding CsA to CyP-D is displaced by Ca2+ [78] implying that under conditions of intracellular Ca2+ overload, CsA might not be able to prevent MPTP opening. In fact, in mitochondria devoid of CyP-D, CsA sensitivity is lost, yet MPTP opening can be seen although at higher Ca2+ [78, 79]. This finding suggests that the effect of CsA represents “desensitization” rather than inhibition of the MPTP [80]. This concept is valid also for all the other available MPTP “inhibitors,” such as Sanglifehrin A and NIM811 [81, 82]. Despite the fact that it is not an MPTP blocker, CsA constitutes an invaluable tool assessing the role of MPTP in myocardial ischemia. Indeed, CsA largely attenuates reperfusion injury. The initial evidence obtained by using a pharmacological approach [13] has now been validated by the development of direct assays for detecting MPTP opening in isolated cells and perfused hearts [83, 84]. Furthermore, MPTP inhibition has recently been shown to limit the loss of viability even when administered only at the time of reperfusion [63]. Definitive support of the role of MPTP desensitization through modulation of its interaction with CyP-D has been provided by the discovery of reduced susceptibility to ischemic injury, observed in mice lacking CyP-D [78, 85]. Altogether, these findings demonstrate that MPTP opening is causally related to the loss of viability and inhibition of this process represents a crucial target for cardioprotection.
Antioxidant and Inhibition of Monoamine Oxidase
Any form antioxidant treatment is potentially cardioprotective at the mitochondrial level based upon the deleterious roles of ROS on mitochondrial function and structure. However, since mitochondria are the main intracellular source of ROS, standard antioxidants do not possess mitochondrial specificity and may diffuse within the cell into other subcellular compartments resulting in a limited efficacy. This serious limitation has prompted the development of the “mitochondriotropic” compounds [86]. The strategy adopted in the design of these molecules resembles that used for assessing mitochondrial membrane potential whereby fluorescent lipophilic cations are electrophoresed into the negatively charged mitochondrial matrix [87]. An example of this strategy is the synthesis of triphenylphosphonium (TPP)-based compounds, which were demonstrated to be effective antioxidants in several cell types [88]. A TPP+-linked analogue of ubiquinone, MitoQ, has decreased reperfusion-induced heart dysfunction, cell death, and mitochondrial damage, whereas unmodified ubiquinone or TPP+ has been not effective [1].
Besides scavenging ROS once they are formed, an alternative and perhaps more efficacious strategy is to prevent their generation or at least the large increase occurring under pathological conditions. This particular goal can be achieved by mild uncoupling. In fact, mitochondrial depolarization decreases ROS generation that is exponentially dependent on Δψm [29]. Decreased ROS formation associated with myocardial protection has been demonstrated by using uncouplers [89] or overexpressing uncoupling proteins [90]. On the other hand, depletion of uncoupling proteins reduces IPC protection and increases ROS formation in cardiac myoblasts [91]. It must be pointed out that a severe uncoupling reduces the loss of viability [92]. This paradoxical finding has been explained as resulting from a decreased availability of ATP for the generation of irreversible hypercontracture, which jeopardizes sarcolemma integrity upon postischemic reperfusion [93, 94]. Another possible mechanism to reduce mitochondrially generated oxidative stress is to inhibit mitochondrial processes generating ROS at sites other than the respiratory chain. For instance, MAO-A has been shown to represent an important source of hydrogen peroxide in rat heart [95], and MAO-A-induced ROS formation resulted in cardiomyocyte apoptosis [96]. Interestingly, a recent study has demonstrated that MAO-A inhibition reduces myocardial damage induced by 30 min of ischemia followed by 60 min of reperfusion in the rat heart [97]. In both isolated cardiomyocytes and in intact heart, MAO-A stimulation has been attributed to an increased availability of its substrate serotonin. Besides investigating whether MAO-A activity is modulated by factors other than an increased supply of exogenous substrates, the specificity of MAO inhibitors should be ascertained by comparing pharmacological evidence with results from a genetic approach based on loss of function studies.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


