Secondary determinants influencing MVO2 include (1) the relative heart rate, (2) myocardial stroke work, (3) imposed afterloads, and/or (4) the inotropic state of the myocardium.
Importantly, associated with cardiac surgery, MVO2 can vary extensively, with the greatest MVO2 occurring immediately after bypass; replenishing energy stores requires a high oxygen demand (i.e., repaying oxygen debt). In contrast, cardiac arrest combined with myocardial hypothermia dramatically reduces MVO2 (Fig. 16.1). It should be noted that hypothermic and normothermic modes of cardiac arrest differ in their degrees of MVO2 reduction. However, in all cases, the arrested heart still elicits an oxygen demand; hence, there will always be some degree of imbalance between oxygen demand and delivery, with ischemia resulting. See Chap. 33 for additional information about cardioprotection during surgery.
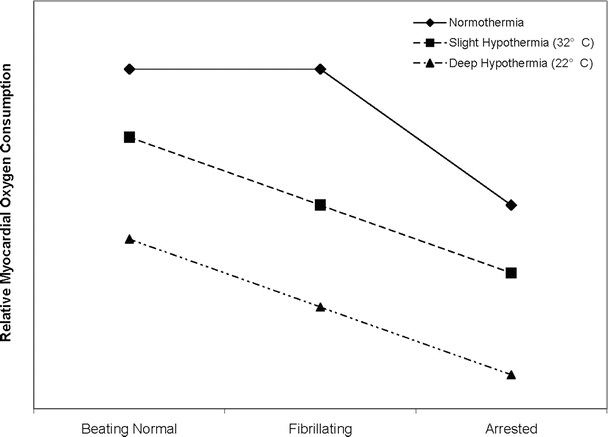
Fig. 16.1
Influence of temperature on myocardial metabolism. While it is expected that hypothermia decreases myocardial oxygen consumption in the beating and fibrillating heart, there also exists a significant difference between the normothermic and hypothermic arrested heart. This indicates that the heart still has a measurable oxygen demand while arrested. Also notable is the difference in myocardial oxygen consumption between the fibrillating and arrested heart at either temperature
16.3 Myocardial Ischemia
The basic definition of myocardial ischemia is a greater myocardial tissue oxygen demand than supply. During short-term ischemic episodes, the heart’s defense mechanism seeks to remedy this imbalance by downregulating myocardial contractile function and, concomitantly, increasing the rate of glycolysis (anaerobic energy production) from preferred lipid metabolism. Consequently, sarcolemmal glucose transport increases, and intracellular acidosis resulting from a buildup of the glycolytic breakdown products causes further inhibition of the contractile apparatus. Even though energy production continues in the absence of oxygen, the glycolytic pathway is an inefficient means for producing ATP. As an ischemic episode becomes more severe or prolonged, the heart becomes unable to produce enough energy via glycolysis and cellular necrosis ensues. For example, 10 min of ischemia generally results in about 50 % depletion of ATP; after approximately 30 min of normothermic ischemia without significant collateral blood flow, irreversible damage or necrosis occurs [1]. Anatomically, the most vulnerable layer of the heart is the subendocardium, due to the higher systolic wall stress in this layer compared to the mid- and epicardial layers; there exists a relatively greater metabolic demand. For additional discussion on myocardial energetics, see Chaps. 15 and 21.
16.4 Ischemic Syndromes
In the past, it was generally believed that extending the periods of myocardial ischemia would, in turn, lead to irreversible damage of the myocardial or infarcted (necrotic) tissue. However, between the clinical conditions of transient ischemia (angina pectoris) and myocardial infarction, five additional ischemic syndromes have been described (Figs. 16.2 and 16.3) [2, 3]. The stunned myocardium is characterized by postischemic impairment of myocardial function, but it is considered acute and completely reversible. The hibernating myocardium is also characterized by depressed myocardial function of variable duration, primarily caused by impaired oxygen delivery through an occluded vessel, and, importantly, recovery of function occurs upon reflow to the ischemic region. Note that the hibernating myocardium is similar to the stunned myocardium, with the main difference being that reperfusion is not the cause of myocardial hibernation, as is the case with myocardial stunning. On the other hand, hibernation can be considered as a state of chronic stunning, yet the exact mechanism of hibernation remains largely unknown [4]. The maimed myocardium is considered the most severe of such syndromes and is characterized by irreversible myocardial damage that follows ischemia and reperfusion and in which there is a delayed recovery to only partial preischemic function. Ischemic preconditioning is the condition where multiple brief ischemic episodes (<5 min) followed by reperfusion subsequently enhance the myocardial tolerances to a longer (<45 min) ischemic event (Figs. 16.3 and 16.4). Finally, patients with ECG changes of ischemia and contractile failure who lack chest pain may be experiencing silent ischemia. It has been proposed that these patients are either less sensitive to painful stimuli or the ischemia is somewhat milder [3].
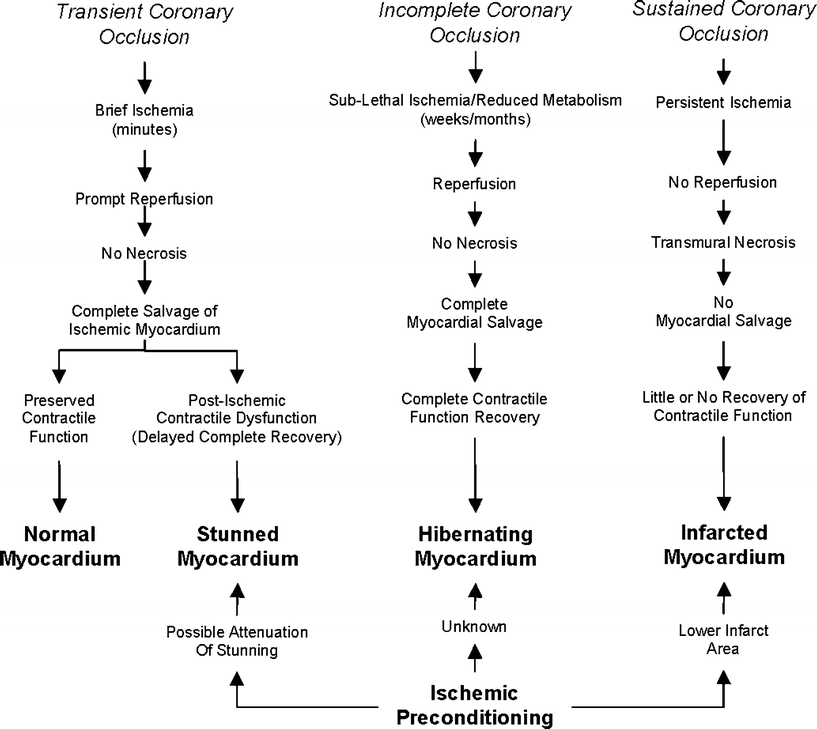
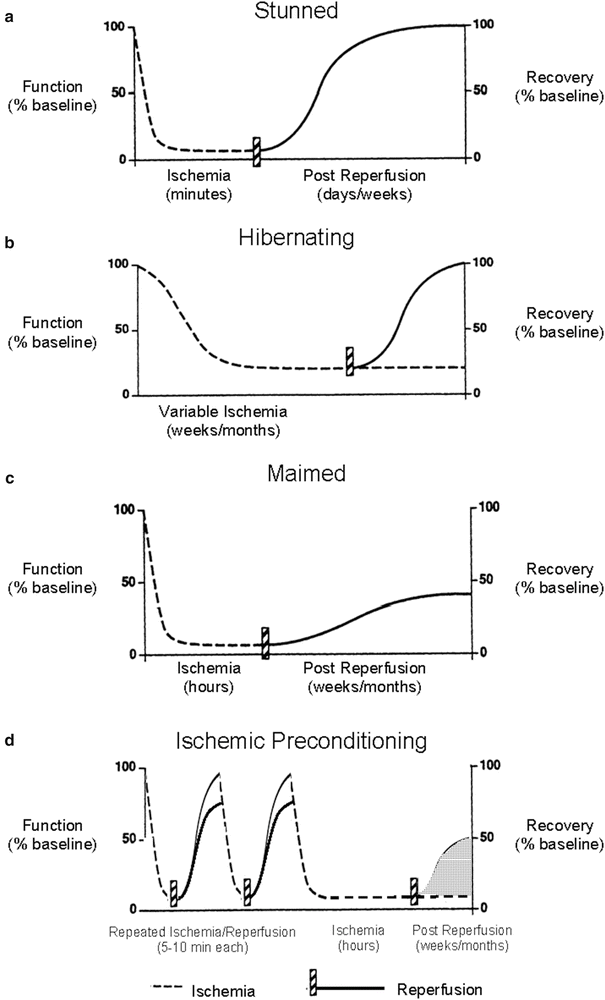
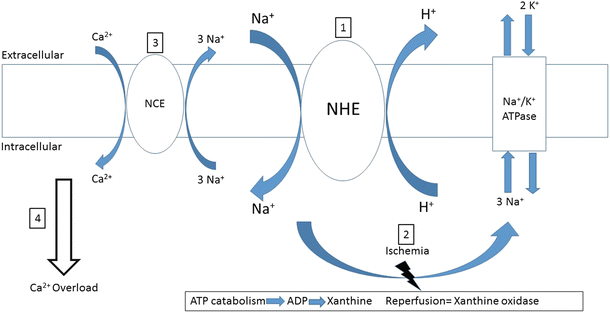
Fig. 16.2
Consequences of myocardial ischemia. The stunned myocardium usually results from a transient coronary occlusion followed by prompt reperfusion; however, it may also occur after prolonged ischemia in the preconditioned heart. Preconditioning lessens the infarct area following a sustained coronary occlusion, but only the area is reperfused. The relationship between preconditioning and the maimed myocardium is unknown. Modified from Boden et al. [17]
Fig. 16.3
New ischemic syndromes. New ischemic syndromes that do not fall within the realm of classic acute reversible and irreversible myocardial ischemia. (a) The stunned myocardium is characterized by a decrease in function following an ischemic event in which there is a complete absence of necrosis from ischemia or reperfusion and a complete functional recovery hours to days later. (b) The hibernating myocardium is characterized by chronic depressed myocardial function due to sublethal ischemia lasting for weeks to months, and revascularization may result in complete recovery of function. (c) The maimed myocardium has permanent damage resulting from a prolonged ischemic episode and has some functional recovery that does not return to preischemic levels. (d) Ischemic preconditioning exists when short ischemic episodes followed by reperfusion confer myocardial protection during a subsequent prolonged ischemic event. However, two areas of uncertainty exist in the preconditioning phenomenon: (1) functional recovery following the preceding short ischemic events may not return to preischemic levels; and (2) while it is known that ischemic preconditioning lessens infarct size, it is uncertain whether long-term functional recovery following the prolonged ischemic episode is significantly improved (via decreased myocardial stunning). Only delayed ischemic preconditioning has been shown to attenuate myocardial stunning. Modified from Boden et al. [17]
Fig. 16.4
Reperfusion injury via the Na+–H+ exchanger. Reperfusion injury-induced calcium overload can be explained, in part, by activation of the Na+–H+ exchanger (NHE). (1) Intracellular acidosis from a prior ischemic episode activates the NHE upon reperfusion, thereby decreasing intracellular acidosis and increasing Na+ influx. (2) Intracellular sodium is primarily removed from the cell via the Na+–K+ATPase during normal myocardial function. However, after ischemia (i.e., during the early stages of reperfusion), the lack of abundance of ATP does not allow for normal operation of the pump and intracellular Na+ increase. (3) Consequently, the Na+–Ca2+ exchanger (NCE), which normally operates by extruding Ca2+ from the cytoplasm, is the primary mechanism for intracellular Na+ removal operating in a reverse mode. (4) Intracellular Ca2+ overload results from NCE activation, possibly causing arrhythmias, stunning, and necrosis
16.4.1 Myocardial Stunning
There are two general theories for explaining the pathomechanisms underlying myocardial stunning, and they are generally not considered to be mutually exclusive. Episodes of ischemia are caused by: (1) the formation of free radical reactive species and/or (2) alterations in intracellular calcium regulation [5]. Furthermore, in such cases, intracellular acidosis occurring during ischemia can potentially generate intracellular calcium oscillations and calcium overload upon reperfusion via activation of the sarcolemmal Na+/H+ exchanger (NHE) (Fig. 16.4) [6].
Experimental studies have shown that the calcium sensitivity of the contractile apparatus is decreased in the stunned myocardium, thereby resulting in lower maximal force generation even at higher than normal transient calcium levels [7, 8]. Furthermore, decreased myofilament sensitivity to calcium is considered primarily responsible for systolic dysfunction; for example, the stunned myocardium is still responsive to inotropic stimulation. Of additional interest is the finding that troponin I (cTnI) degradation products were discovered in the human myocardium during aortic cross-clamping with bypass and that serum levels of cTnI increased during reperfusion, peaking approximately 24 h following cross-clamp removal [9]. This preliminary evidence further suggests that cTnI degradation products may potentially be utilized as biomarkers for occurrence of myocardial stunning.
During the early stage of stunning, it is considered beneficial to prevent calcium oscillations and thus attenuate significant injury caused by reperfusion. This has been accomplished experimentally by utilizing Ca2+ antagonists, inorganic blockers, ryanodine, low-calcium reperfusion buffers, and/or NHE blockers [10]. Conversely, when contractility is suppressed, as in the late stage of stunning, therapies should include those that increase the amplitudes of intracellular calcium transients, inducing inotropic responses. Included in this subset of therapeutics are high calcium buffers, Ca2+ agonists, catecholamines, and/or phosphodiesterase inhibitors. Importantly, many of these therapies are specific to the stage or degree of stunning, and hence, the timing of their use is critical, so as not to become a detrimental therapy. For instance, hypocalcemia during and following cardiopulmonary bypass is a common occurrence mainly attributed to the utilization of priming fluids, citrated blood, and large doses of heparin during bypass [11]. Normally, hypocalcemia is successfully corrected with the administration of calcium chloride; however, calcium levels may return to preoperative levels prior to removal from cardiopulmonary bypass without supplemental calcium [12]. Therefore, the risk of stunning and myocardial damage may actually be increased with generalized calcium chloride administration. While it may be used for post-bypass cardiac resuscitation, current Advanced Cardiac Life Support (ACLS) guidelines do not indicate calcium chloride for general resuscitation purposes, and administration if done at all is on an “as necessary” basis [13].
16.4.2 Hibernating Myocardium
As previously discussed, myocardial stunning and hibernation are related in that a depressed state of contractility exists and, yet importantly, there is the potential to reversibly return the dysfunctional myocardium back to normal. However, it must again be restated that while stunning can be attributed to the reperfusion following a brief bout of ischemia, the hibernating myocardium is in a chronic hypocontractile state, due to a decreased oxygen supply and thus may only recover full function with revascularization therapy. In other words, this reduction in oxygen delivery is attributable to reductions in local perfusion, and the corresponding reduction in contractility has been shown to be a proportional response of the myocardium, termed perfusion-contraction matching and elucidated by John Ross [14]. It may be beneficial to consider the hibernating myocardium as a viable tissue which is simply eliciting necessary physiological compromises in response to locally limited blood flow. Additionally, many of the underlying mechanisms of stunning are considered directly related to the detrimental effects of reperfusion injury (to be discussed later) [15]. While there is a general absence of necrosis with myocardial hibernation, morphological changes to the myocardial architecture, such as loss of myofibrils and increased interstitial fibrosis, may occur if this state persists [16].
16.4.3 Maimed Myocardium
The maimed myocardium closely resembles that of a heart with a classic myocardial infarction in that ischemia-induced necrosis leads to loss of contractile performance. In these cases, unlike in myocardial stunning, the duration of ischemia is long enough to result in necrosis. Importantly there can be partial recovery of function in this ischemic region, i.e., following timely reperfusion [17]. An example of the maimed myocardium syndrome would be a patient that exhibits an incomplete recovery of myocardial function following drug-induced or mechanical (angioplasty) reperfusion of an occluded coronary artery and then subsequently demonstrates regions of viable myocardium in the original ischemic area.
16.4.4 Ischemic Preconditioning
Ischemic preconditioning is a biological phenomenon whereby brief ischemic episodes followed by adequate reperfusion protect tissue from a subsequent prolonged ischemic event [18]. In general, cardiac protection from ischemic preconditioning has been characterized as biphasic. The first window of protection occurs immediately following the ischemic event for approximately 3 h mediated by effectors. The second window of protection occurs some 24 h after the ischemic event and can last for up to 3 days and is mediated by an upregulation of cardioprotective proteins. There are several signaling pathways that are involved in these mechanisms of ischemic preconditioning, including the activation of potassium channels that are located in the inner membrane of the mitochondria of the cardiac myocytes [19]. These pathways can be utilized as drug delivery targets for the therapeutic treatment of prevention for ischemic injury, by mimicking the natural progression of limited ischemic preconditioning. In the myocardium, ischemic preconditioning has been shown to be potentially infarct limiting [18] as well as antiarrhythmic [20], although the latter of these effects can be agent specific. It is also well established that endogenous opioid receptor activation participates in myocardial ischemic preconditioning [21] and that preischemic administration of synthetic opioid agonists can mimic the benefits of ischemic preconditioning [22]. While ischemic and opioid preconditioning have both been convincingly shown to delay cell death in various experimental animal models, the clinical applicability of these therapies in humans may be limited to situations where the ischemic event can be anticipated (e.g., on- or off-bypass cardiac surgery, percutaneous transluminal coronary angioplasty, or stenting procedures) [23]. For a more comprehensive review of ischemic and opioid preconditioning, see articles by Yellon and Downey [24] (ischemic preconditioning) and Gross [25] (opioid preconditioning).
16.4.5 Silent Ischemia
Silent ischemia refers to single or multiple asymptomatic episodes of transient ischemia. In some cases, silent ischemia can occur in the week(s) following an acute myocardial infarction in patients with a history of coronary artery disease. In other scenarios, seemingly normal healthy individuals may experience episodes of silent ischemia that go relatively unnoticed. Detection of silent ischemia primarily relies on electrocardiographic monitoring, either in the form of 24-h Holter monitoring (ambulatory) or exercise- and/or stress-induced assessment. In both cases, ischemia is typically detected by asymptomatic ST-segment elevations in an ECG tracing [26]; see also Chap. 19. Silent ischemia is postulated to be related to a lack of oxygen supply rather than an increase in oxygen demand; however, controversy remains regarding the mechanisms whereby silent ischemia can proceed unnoticed.
16.4.6 How Can the Heart Be Protected from Ischemia?
As shown in Fig. 16.1, there are several means of decreasing myocardial oxygen demand when the oxygen supply is compromised. These include hypothermia, pharmacologically decreasing the heart rate, and/or controlled cardiac arrest. Additionally, as mentioned above, ischemic and pharmacological preconditioning of the heart are other ways to protect the myocardium from an ischemic episode and, hence, induce cardioprotection. However, these therapies require the anticipation of the ischemic event, such that treatment can be administered prior to or early during the ischemia. In situations where anticipation of ischemia is not possible, for example, in acute myocardial infarction, interventions which reestablish blood flow and oxygen to the ischemic zone (reperfusion) are the primary therapies. Nevertheless, it should be noted that dietary supplements such as omega-3 fatty acids have been considered as a potential means for prevention [27, 28].
16.5 Reperfusion Injury
While immediate restoration of blood flow and oxygen to ischemic tissue is ultimately a beneficial and important therapy, it should be noted that additional myocardial damage can occur with such myocardial reperfusion itself. In what has been termed the oxygen paradox, the resupply of oxygen to a hypoxic cell simultaneously activates two intracellular processes of particular interest—the membrane-bound calcium pumps and the contractile apparatus. Resumption of contractile activity in the presence of oscillating and increasing intracellular Ca2+ levels can force the heart into a state of hypercontracture and thus cause intracellular edema.
Another proposed mechanism of injury during reperfusion can result from xanthine oxidase. During periods of ischemia, ATP is catabolized and ATP is reduced to ADP, AMP, adenosine, and inosine and further reduced to xanthine. Xanthine alone is not cytotoxic, but when oxygen is reintroduced, the product is xanthine oxidase. This in turn releases free radicals into the myocardial tissue via super oxide dismutase, hydrogen peroxide, and uric acid [29]. Hence, the administration of xanthine oxidase inhibitors can diminish the effects of free radical production when a procedure is planned. For example, a patient could be given prophylactic xanthine oxidase inhibitors prior to a coronary artery bypass graft when reperfusion injury is anticipated. Collectively, these etiologies may ultimately result in membrane disruption and even cell death.
Reperfusion injury can present in (or is associated with) one or more of the following pathologies: (1) reperfusion arrhythmias, (2) microvascular damage and no-reflow, (3) accelerated cell death, (4) myocardial stunning, and/or (5) post-pump syndrome in procedures requiring cardiopulmonary bypass (Fig. 16.5). Further, reperfusion injury may cause immediate myocardial necrosis in severely damaged cells and delayed necrosis in cells adjacent to the ischemic region; conversely, complete recovery of myocardial function may occur despite an ischemic episode. Of importance, necrosis occurring during the ischemic episode must be differentiated from that which may occur following reperfusion, especially when discussing clinical therapies targeted at attenuating reperfusion injury.
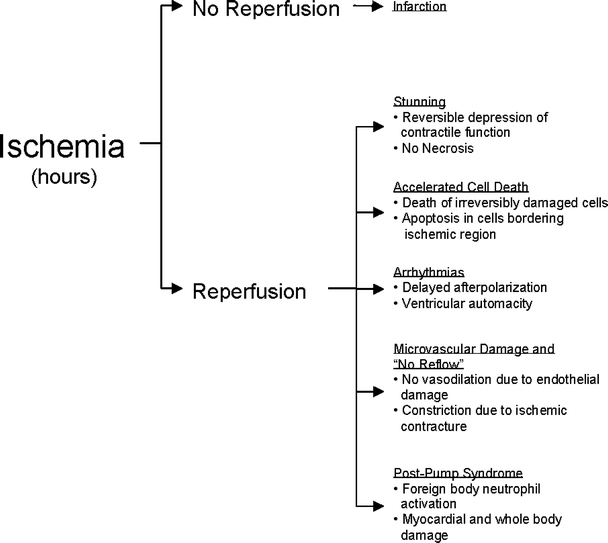
Fig. 16.5
Aspects of reperfusion injury. While reperfusion remains the most beneficial therapy for ischemia, any combination of stunning, accelerated cell death, arrhythmias, microvascular damage, or post-pump syndrome could occur, thus leading to postischemic dysfunction or necrosis
Assessment of reperfusion injury following ischemia is often difficult, especially in postsurgical patients. Yet, the determination of reperfusion injury and the relative extent of injury can be indirectly accomplished by hemodynamic monitoring (pressures, cardiac outputs, and echocardiography) and examination of blood levels of cardiac enzymes (CK-MB, troponin I, LDH, and AST). Ideally, left ventricular end-diastolic pressure volume measurements will provide both functional and quantitative information relative to the degree of reperfusion injury; yet, such data are difficult and rarely feasible to obtain clinically. With recent advances in echocardiography, relative changes in cardiac function and regional wall motion can be assessed more readily in such patients (see Chap. 22). Furthermore, improvements in cardiac magnetic resonance imaging have allowed for the functional mapping of ischemic myocardial zones (see Chap. 24).
Myocardial viability can be fairly easily assessed with inotropic stimulation, as the postischemic stunned (or the potentially reversibly injured) myocardium will display an increased heart rate and contractility. In contrast, the irreversibly injured (necrotic) myocardium exhibits little to no response to the inotrope (e.g., by dopamine stress echocardiography). Note that, by definition, myocardial stunning is reversible; therefore, within days, a depressed cardiac function due to stunning should recover (Figs. 16.2 and 16.3). This phenomenon is commonly observed clinically when patients following coronary artery bypass grafting require 24–48 h of inotropic support.
16.5.1 Aspects of Reperfusion Injury
16.5.1.1 Myocardial Stunning
The presence of intracellular oxygen free radicals and increased intracellular calcium during reperfusion leads to a reversible hypocontractile state of variability, yet this is relatively brief in duration.
16.5.1.2 Accelerated Cell Death
Typically, accelerated cell death upon reperfusion refers to cells that have been irreversibly damaged during the prior ischemic episode and are destined to die despite reperfusion. However, irreversible damage is not a prerequisite for cell death; upon reperfusion, detrimental ischemia-induced intracellular alterations may also occur in viable cells. More specifically, during reperfusion, the development of increased sarcolemmal permeability due to ischemia allows for the uncontrolled influx of calcium resulting in hypercontracture, decreased energy production, and/or cell death. Additionally, it should be noted that there is the paradoxical finding that apoptosis-related cell death in postischemic viable myocardium is reduced by early reperfusion and is accelerated in irreversibly ischemic-damaged cells [30].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


