Restenosis
Heba Wassif MD, MPH
Frederick GP Welt MD
Endovascular mechanical therapy for vascular stenosis was first described by Dotter and Judkins (1). Fourteen years later, in 1978, Andreas Gruentzig published the results of the first percutaneous transluminal coronary balloon angioplasty (PTCA) in a landmark study of a series of five patients (2). Yet, despite the revolutionary impact of this procedure on the treatment of coronary artery disease, even within this initial publication, Gruentzig acknowledged a rate of clinical restenosis of ˜19% (3). In the past 35 years, interventional cardiology has witnessed vast improvements designed to eliminate some of the limitations of balloon angioplasty. However, despite the advent of effective therapies, postintervention restenosis remains an important limitation of treatment with coronary stents. The introduction of bare metal stents (BMSs) decreased the in-stent restenosis (ISR) rate compared with the balloon angioplasty rate, yet it remained as high as 30% (4). Further introduction of drug-eluting stents (DESs) reduced the rates to 5% to 18%, depending on the population (5, 6, 7 and 8). The understanding of the cellular and molecular mechanisms that contribute to restenosis remains key in introducing therapies. Marked improvement in this understanding has accumulated over the past decade, and the field remains active.
The aim of this chapter is to understand the pathogenesis of restenosis, the clinical indicators, and the biochemical markers that are associated with increased risk, and to introduce the evidence behind therapy to date.
DEFINITIONS AND CLINICAL SEQUELAE OF RESTENOSIS
For most clinical trials, restenosis is defined by the binary endpoint of a reduction of 50% of the luminal diameter compared with the reference vessel (9). This is somewhat of an arbitrary definition that endeavors to separate clinically important lesions from those without sequelae. Restenosis can also be described in more continuous terms, which are helpful in understanding the pathophysiology of the process. As an example, late lumen loss (LLL) is defined as the continuous angiographic measure of lumen deterioration, and calculated by subtracting minimal lumen diameter (MLD) at immediate follow-up from late postprocedural MLD. This parameter has been used as a surrogate marker for the effects of different antirestenotic therapies and to determine outcomes in the BMS era and continued in the DES era (10, 11). One important point derived from viewing restensosis as a continuous variable is the fundamental understanding that virtually all patients suffer some degree of restenosis, which follows a typical Gaussian distribution (Fig. 2-1). However, only the few patients who have severe ISR (usually with a lesion severity of >70%) have symptoms. The average amount of LLL is 0.8 to 1.0 mm after implantation of a BMS. This amount of late loss occurs regardless of the size of the original vessel lumen. This is of critical significance, especially for smaller vessels. A 4-mm-diameter vessel would lose 44% of lumen area with the loss of 1-mm diameter. If a vessel with a 2-mm diameter experiences the same 1 mm of late loss, there is a 75% loss of lumen area and angiographic restenosis. Therefore, late loss and angiographic restenosis are related to the diameter of the reference vessel (12). One of the most important concepts to understand is that restenosis rates vary hugely, depending on the particular definition used in any given study (Table 2-1).
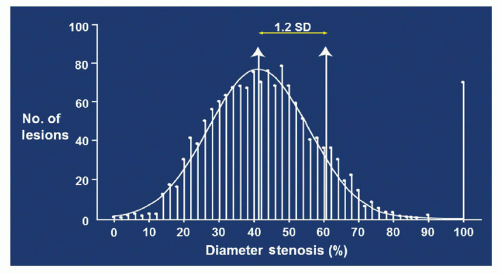 FIGURE 2-1 Histogram of percent diameter stenosis at follow-up angiography of 1,445 lesions treated with percutaneous intervention. The superimposed curve represents the theoretic Gaussian distribution. (From: Rensing BJ, et al. J Am Coll Cardiol. 1992;19:939-945, with permission.) |
Discordance between late loss and binary restenosis has been observed in several trials and is probably multifactorial and has been a topic of much debate. Although late loss has been very useful as a surrogate marker for the effectiveness of a given drug to prevent clinical restenosis, the relationship between late loss and binary restenosis is nonlinear and has been characterized by a curvilinear function. This notion implies that there is a threshold for clinical significance. The argument is that below a given amount of late loss, the degree of tissue growth is immaterial as it does not reflect a worse clinical restenosis rate (Fig. 2-2). This argument is not universally supported.
TABLE 2-1 Definitions of Restenosis Used in the Literature | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The clinical sequelae of angiographic restenosis are not always in concordance with the degree of restenosis. In a meta-analysis of all patients with angiographic ISR from the BENESTENT I, BENESTENT II pilot, BENESTENT II, MUSIC, WEST 1, DUET, FINESS 2, Fluvastatin Angioplasty Restenosis (FLARE), SOPHOS, and ROSE studies, which recruited 2,690 patients who underwent percutaneous revascularization, restenosis (≥50% diameter stenosis) occurred in 607 patients and was clinically silent in almost half those patients. There are likely multiple factors that play a role in how patients present, including reference diameter and lesion severity, in follow-up (13).
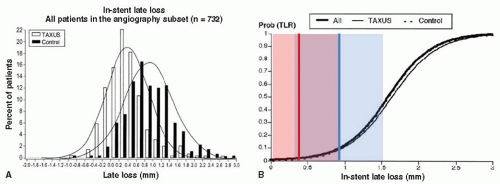 FIGURE 2-2 Data from the TAXUS IV trial show the in-stent late loss for Taxus and control bare metal stent in panel A. Panel B shows the probability of target lesion revascularization (TLR) as a function of in-stent late loss revealing a curvilinear distribution. Superimposed are the mean and standard deviations for the Taxus stent late loss (red) and the control bare metal stent (blue). The distribution of the Taxus stent late loss falls along the relatively flat portion of the curve, whereas the distribution for the bare metal stent falls along the steeper portion of the curve, where there is greater correlation between late loss and TLR. (From: Ellis et al. J Am Coll Cardiol. 2005;45:1193-1200, with permission.) |
Traditionally, restenosis has been viewed as a largely benign condition presenting most commonly with stable angina. However, there is substantial evidence to the contrary. Bare metal ISR had shown variability in presentation. Although the majority present with stable exertional symptoms, unstable angina is a frequent presentation (26%-53%), and acute myocardial infarction (MI) has been described in 3.5% to 20% of patients (14, 15). DESs have similar presentations. In one series, the rates of unstable angina and MI were comparable between the two groups, 18% and 2%, respectively (16). These stud ies point to the fact that ISR is not always a benign condition.
One of the special concerns regarding restenosis is the potential greater risk in the setting of left main stenting, which has been increasing in frequency in recent years, and, though it remains <1% of percutaneous coronary intervention (PCI) in the United States, is likely to increase. Despite concerns regarding the possible serious consequences of restenosis in this scenario, multiple case series such as the Rotterdam and Milan series have shown promising results of left main stenting compared with coronary artery bypass surgery. The safety of the procedure has also been well documented in randomized trials, and follow-up angiography is no longer routinely recommended after the procedure (17, 18 and 19).
Multiple studies have confirmed that compared with atherosclerosis, restenosis following balloon angioplasty or stent implantation is a relatively accelerated process. With balloon angioplasty, clinically important restenosis is almost always apparent by 6 months (20, 21). Similarly, in stents, the average time at which ISR is apparent is at 5.5 months, with some evidence that there is a shorter interval if presentation was with acute MI (22).
RISK FACTORS FOR RESTENOSIS
The three most important clinical risk factors for restenosis that have been identified are increased stent length, smaller MLD, and the presence of diabetes (23). Risk factors can be loosely identified as both biologic in nature and mechanical, although there is considerable interplay between the two.
Biologic
Risk for restenosis is particularly high among patients with diabetes mellitus. Data from the Swedish Coronary Angiography and Angioplasty Registry (SCAAR), registry involving >35,000 patients implanted with four different types of DESs (Endeavor, sirolimus-eluting stents (SES), Taxus Express, and Liberte) in real-world practice at 2-year follow-up, showed that the rates of restenosis with DES implantation were significantly higher in patients with diabetes (24).
In a another study of 954 patients undergoing PCI, target lesion revascularization (TLR) was required in 28% of patients with insulin-dependent diabetes compared with 16.3% of individuals without diabetes (25). The high risk for restenosis among patients with diabetes may be associated with metabolic alterations that promote endothelial dysfunction, accelerate intimal hyperplasia, and increase platelet aggregability and thrombogenicity (26).
Evidence also suggests that some patients may be genetically predisposed to restenosis. Genetic abnormalities associated with high risk for restenosis include polymorphisms in genes coding for angiotensin-converting enzyme (ACE) inhibitor (27), glycoprotein (GP) receptor IIIa PLA1/2, haptoglobin 2/2.25, and IL8 (28). Resistance to antiproliferative drugs due to polymorphisms in the genes that encode the intracellular receptor mammalian target of rapamycin (mTOR) or proteins involved in paclitaxel or sirolimus metabolism have been shown to confer drug resistance both in vitro and in vivo (29, 30). Decreased binding of sirolimus to mTOR because of mutations in FK-B12 and mTOR, and mutations of downstream effector molecules of mTOR may all cause resistance to sirolimus (30).
Attempts have been made to identify markers of inflammation and thrombosis that are correlated with increased risk of ISR. Local markers include a strong correlation between the number of macrophages in tissue samples obtained at the time of directional atherectomy and the propensity for restenois (31). However, obtaining pathologic specimens is neither feasible nor practical, and investigators have pursued local and systemic biomarkers that could assist in identifying patients and lesions at high risk of restenosis. Neumann et al. opted to obtain blood samples proximal and distal to the site of balloon angioplasty in humans. They determined the expression of neutrophil adhesion molecule L selectin and CD 11b, and found there was upregulation of markers following balloon angioplasty (32). Mickelson used systemic venous blood sampling for measurement of CD11b and found similar results, with propensity toward adverse clinical events (33). Monocyte chemoattractant protein-1 (MCP-1) is a potent chemoattractant of monocytes. Plasma levels of MCP-1 were measured before and 1, 5, 15, and 180 days after PTCA in 50 patients who underwent PTCA and who had repeated angiograms at 6-month follow-up. Restenosis occurred in 14 (28%) patients. The MCP-1 level was no different at baseline between patients with or without restenosis. However, after the procedure, restenotic patients, compared with nonrestenotic patients, had statistically significant (p < 0.0001) elevated levels of MCP-1. In contrast, plasma levels of other chemokines, such as RANTES and interleukin-8, did not differ between the two groups after PTCA. Higher MCP-1 throughout the study was correlated with restenosis (34).
The plasma level of C-reactive protein (CRP) is considered a risk predictor for cardiovascular (CV) diseases. However, the relationship between CRP and ISR has been controversial. There have been studies that supported the notion that increased inflammation following PCI is higher with BMSs than with DESs conferred by lower CRP levels periprocedural with DESs (35, 36), which conflicted with other studies that showed higher levels of CRP. When the goal of the study was to investigate whether CRP was linked to restenosis, the results were mixed (37, 38 and 39).
Circulating matrix metalloproteinases (MMPs) recently have been demonstrated to be potentially useful in identifying patients at greater risk of developing ISR following DES implantation. MMP-2 and MMP-9 are well known to play fundamental roles in the migration of vascular smooth muscle cells (VSMCs) and matrix remodeling during wound healing, and are produced by VSMCs, endothelial cells, macrophages, lymphocytes, and mast cells in response to mechanical injury. Significant elevations in MMP-9 levels at baseline and in MMP-2 and MMP-9 levels 24 hours after PCI have proven to be strongly associated with the development of ISR following DES implantation. Conversely, low and near-normal MMP-2 and MMP-9 levels were associated with a lack of a significant restenotic response (40).
Mechanical
Stent underexpansion and issues of malapposition (both early and late) are factors more predisposed to stent thrombosis than to increased restenosis, although some authors have suggested that incomplete expansion also impacts drug delivery and subsequently enhances neointimal hyperplasia (NIH) (41, 42). In the era of DESs, the concept of geographical miss was introduced, referring to an area of the treated segment that was exposed to balloon injury but not covered with stent struts and therefore not receiving adequate suppressive therapy. Less common is stent fracture, defined as complete or partial separation of a stent at follow-up that was contagious after the original stent implantation (43). The incidence of stent fracture with DESs has been reported as 1% to 8% (44, 45 and 46). With stent fracture, the scaffolding is impaired and subsequently drug delivery may be impacted (47).
PATTERNS OF ISR
Mehran et al. developed an angiographic classification of ISR according to the geographic distribution of intimal hyperplasia in reference to the implanted stent (48). That included four classes defined as the following (Fig. 2-3):
Class I: Focal ISR group—Lesions are 10 mm in length and are positioned at the unscaffolded segment (i.e., articulation or gap), the body of the stent, the proximal or distal margin (but not both), or a combination of these sites (multifocal ISR).
Class II: “Diffuse intrastent” ISR—Lesions are 10 mm in length and are confined to the stent(s), without extending outside the margins of the stent(s).
Class III: “Diffuse proliferative” ISR—Lesions are 10 mm in length and extend beyond the margin(s) of the stent(s).
Class IV: ISR with “total occlusion”—Lesions have a thrombolysis in myocardial ischemia (TIMI) flow grade of 0.
PATHOGENESIS OF RESTENOSIS
The pathogenesis of restenosis is significantly different from atherogenesis in both the content of the lesion and the time required for development. As discussed above, restenosis develops much more
rapidly than atherosclerosis and is sometimes referred to as an accelerated arteriopathy. Forrester proposed a paradigm for restenosis based on the vascular biology of wound healing and suggested three phases in the process: the inflammatory phase, a granulation or cellular proliferation phase, and a phase of remodeling involving extracellular matrix (ECM) protein synthesis (49). More recently, Park et al. have suggested a possible occurrence of in-stent neoatherosclerosis in selected situations, although the prevalence of these histologic findings is uncertain (50).
rapidly than atherosclerosis and is sometimes referred to as an accelerated arteriopathy. Forrester proposed a paradigm for restenosis based on the vascular biology of wound healing and suggested three phases in the process: the inflammatory phase, a granulation or cellular proliferation phase, and a phase of remodeling involving extracellular matrix (ECM) protein synthesis (49). More recently, Park et al. have suggested a possible occurrence of in-stent neoatherosclerosis in selected situations, although the prevalence of these histologic findings is uncertain (50).
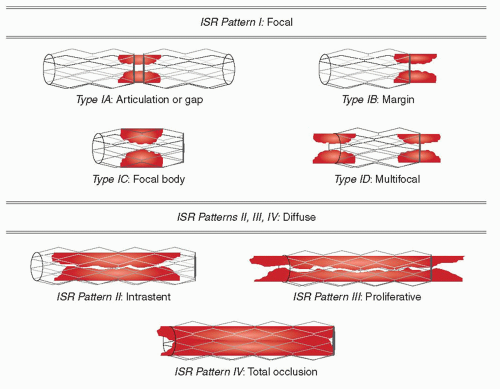 FIGURE 2-3 Common patterns of in-stent restenosis. (Redrawn from: Mehran et al. Circulation. 1999;100(18):1872-1878, with permission.) |
DIFFERENCES BETWEEN POST-BALLOON ANGIOPLASTY AND ISR
Before discussing the molecular and cellular mechanisms of restenosis, it is worth elucidating the important differences that exist between restenosis following balloon angioplasty alone and in-stents.
Balloon Angioplasty
Detailed intravascular ultrasound (IVUS) studies have helped to identify the mechanisms involved in postangioplasty restenosis. These studies demonstrate that restenosis is caused both by neointimal proliferation and shrinkage of the artery. Elastic recoil is an acute process that is observed within a few minutes after balloon deflation, resulting in inward collapse of the vessel wall. More stretching leads to increased inward recoil force upon balloon inflation. The recoil force can result in up to 50% loss of cross-sectional area and a 33% loss in luminal diameter (51). However, negative remodeling recognized as contraction of the external elastic laminae is a process that typically presents weeks to months after injury. Neointimal proliferation is considered an inflammatory response (discussed below) and is thought to result from inflammation at the site of injury, smooth muscle cell (SMC) proliferation and migration, and excessive ECM production. These last two processes contribute to neointima (NI) formation (52, 53). In terms of their relative contribution, it has been demonstrated that acute and chronic negative remodeling play the larger role in balloon-injury-associated restenosis (Fig. 2-4).
 FIGURE 2-4 Illustration of differences in mechanisms of restenosis between plain balloon angioplasty and stenting. In balloon-angioplastied vessels, restenosis is caused by a combination of neointimal growth and negative remodeling. Stented arteries have lower rates of restenosis despite incurring greater neointimal growth because of their ability to achieve a larger initial lumen size, and the elimination of negative remodeling. |
In-Stent Restenosis
Angiographic analysis of the pivotal studies that introduced coronary stents (the Stent Restenosis Study [STRESS] and the Belgian Netherlands Stent Study [BENESTENT]) revealed major quantitative and qualitative differences between angioplasty-induced injury and that associated with stent placement. The initial luminal gain with stents is greater due to scaffolding of the vessel. In addition, late negative remodeling is abrogated by the presence of the rigid frame of the stent. However, the late luminal loss is greater with stented vessels (Fig. 2-4). It is explained on the basis of greater neointimal growth that erodes into the primary gain (54, 55). Hoffman et al. compared stented and nonstented lesions using serial IVUS studies, confirming the observation that the main mechanism of ISR is due to NIH rather than negative remodeling (56). Therefore, stents were shown to reduce restenosis by the fact that they are able to achieve a larger initial size of the lumen and prevent late remodeling. However, they actually induce more late loss.
The neointimal formation that occurs in ISR is similar to that following balloon angioplasty. The exaggerated response seen with stenting is likely secondary to the differences in vessel injury and more potent inflammation. Kornowski et al. demonstrated, using a porcine model, that an increase in vessel injury or vascular inflammation results in an increase in neointimal formation. Pigs in which the stent struts perforated the internal elastic lamina and external elastic lamina had greater histologic evidence of inflammatory response and subsequently a larger volume of neointimal formation (57). Other contributing factors to increased inflammatory response may be derived from the increased balloon pressure required to place the stent and contact allergy. Contact allergy to metals, including nickel and molybdenum, may account for the elevated inflammatory response observed with stents in some patients. BMSs slowly elute metal ions that may stimulate a delayed-type hypersensitivity response within the stented vessel (58). A study was conducted in Germany on 131 patients who underwent cutaneous patch testing after stent implantation to investigate the association of nickel and molybdenum hypersensitivity with ISR. All patients with a positive test result (n = 10) had restenosis (p = 0.03), requiring target vessel revascularization (TVR) (58). Although the number of patients evaluated in studies investigating the correlation between metal allergy and ISR is small, these elucidating data suggest that metallic hypersensitivity may account for a subset of restenosis observed with bare-metal stenting.
INFLAMMATION MECHANISMS OF LEUKOCYTE RECRUITMENT AND INFILTRATION
Expansion of a balloon or placement of a stent causes injury within the vessel wall, including dissection, crush injury of SMCs, and de-endothelialization. Leukocyte recruitment and infiltration occur at these sites of injury, where platelets and fibrin have been deposited. Within areas of injury such as atherosclerotic and postangioplasty restenotic lesions, and in areas of ischemia-reperfusion injury, in vivo studies have shown that leukocytes and platelets are deposited together. For the inflammatory response after angioplasty, this interaction between platelets and leukocytes appears to be important (59).
This interaction has been explained by Diacovo et al., who have put forth a paradigm of leukocyte attachment to surface-adherent platelets followed by transmigration (60). As with atherosclerosis, the initial loose association of leukocytes is mediated through the selectin class of adhesion molecules, particularly by platelet P-selectin, followed by their firm adhesion and transplatelet migration, processes that are dependent on the integrin class of adhesion molecules (60, 61). The β2 integrin molecule Mac-1 (CD11b/CD18) is present on both neutrophils and monocytes, and appears to be of central importance in leukocyte recruitment following vascular injury. In addition to promoting the accumulation of leukocytes at sites of vascular injury, the binding of platelets to neutrophils amplifies the inflammatory response by inducing neutrophil activation, upregulating cell-adhesion-molecule expression, and generating signals that promote integrin activation and chemokine synthesis. These processes of activation may be mediated through the release of soluble CD40 ligand, a proinflam-matory molecule stored most abundantly in platelets. Bolstering these data, both neutrophil-platelet and monocyte-platelet aggregates have been identified in the peripheral blood of patients with coronary artery disease, and may be markers of disease activity and prognosis (62, 63).
EVIDENCE FOR A ROLE OF INFLAMMATION IN RESTENOSIS
In most cases, restenosis does not appear to be a case of accelerated atherosclerosis, but rather a distinct temporal and pathophysiologic process. However, inflammation is an important common link between atherogenesis and restenosis. Observations regarding restenosis have been hampered by the difficulty of obtaining human restenotic tissue. Farb et al. investigated stented arteries from pathologic samples of 116 stents from 87 patients >90 days post procedure. They found a statistically significant association between extent of medial damage, inflammation, and restenosis (64). Also linking leukocytes and restenosis is a study by Moreno et al., in which the authors present data from tissue retrieved from directional atherectomy at the time of angioplasty. They found a strong positive correlation between the number of macrophages present in the tissue at the time of angioplasty and subsequent risk of restenosis (65).
Systemic markers of inflammation following angioplasty have also provided insight into the mechanisms of restenosis. Neumann et al. investigated systemic inflammation by collecting blood samples both proximal to and just distal to the site of balloon dilatation in humans. Using flow cytometry to determine the expression of the neutrophil adhesion molecules L-selectin and CD11b, they found an upregulation of these markers of leukocyte activation after angioplasty, measured as a gradient of distal and proximal specimens (32). Mickelson et al. were able to document upregulation of CD11b on both neutrophils and monocytes using systemic venous specimens from patients undergoing angioplasty. They found that levels of CD11b had a positive correlation with adverse clinical events (33). Inoue et al. confirmed these data, demonstrating that elevated levels of neutrophil CD11b are predictive of future propensity for restenosis after balloon angioplasty and stenting (66, 67). Pietersma et al. showed that interleukin 1 production by stimulated monocytes isolated from blood before angioplasty was correlated positively with late luminal loss, while activation of granulocytes measured by CD66 levels was inversely correlated with late loss (68). Cipollone et al. demonstrated upregulated levels of MCP-1 following percutaneous intervention in humans, and found that MCP-1 levels correlate with risk for restenosis (34). Gaspardone et al. showed a positive correlation between the nonspecific inflammatory marker CRP following stent placement and propensity for restenosis (69).
Animal models have been invaluable in elucidating basic cellular and molecular mechanisms of restenosis. In several experimental animal models, cell adhesion molecules critical for leukocyte recruitment have been found to be upregulated by an atherogenic diet (70




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
